 ISSN
ISSN 


-
药物开发周期长、耗资大,药物流失率高。目前,每10个候选药物中就有9个在I期临床试验或监管批准时失败[1]。为改善药物发现过程效率低下的状况,缩短新药研发周期及提高成功率,药物化学家们提出了定量构效关系(quantitative structure - activity relationships, QSAR)的概念。QSAR是对已知先导化合物的一系列衍生物进行定量的生物活性测定,分析衍生物的理化参数与生物活性的关系,建立结构与生物活性之间的数学模型,并以这种数学模型来指导药物分子设计[2]。早期阶段,机器学习方法是QSAR领域较为常用的建模方法。由于传统机器学习方法只能处理固定大小的输入,大多早期的QSAR建模都是针对不同任务,人工生成相应的固定长度的分子描述符。常用的分子描述符包括[3]:1)分子指纹,通过一系列表示特定子结构的二进制数字对分子结构进行编码[3];2)一维/二维分子描述符:由统计学家和化学家处理的描述分子物理化学和微分拓扑衍生的描述符[3]。常用的建模方法包括线性方法(如线性回归)和非线性方法(如支持向量机、随机森林等)。近年来,深度学习方法已成为QSAR建模的最新研究方向。
过去十年中,深度学习已成为各领域的主要建模方法,尤其在医学领域,涉及生物活性预测、药物从头设计、医学图像分析和合成预测等多个方向。卷积神经网络(convolutional neural networks, CNN)是深度学习中的一种特殊架构,已成功解决了结构化数据(如图像)的问题[4]。但是,当图形具有不规则形状和大小、节点位置没有空间顺序且节点的邻居也与位置有关时,传统卷积神经网络则不能直接应用于图上。针对这种非欧式结构化数据,研究者们提出了图卷积网络(graph convolutional network, GCN),且基于此提出了各种衍生架构。文献[5]提出了第一个图神经网络(graph neural networks, GNN),该架构基于递归神经网络学习了无向图、有向图和循环图的体系结构。文献[6]基于频谱图理论提出了图卷积网络。目前,已有其他形式的GCN,如图注意网络(graph attention network, GAT)、图自动编码器和时空图卷积等。
近几年,已有多数研究将图卷积应用于分子的生物活性预测。在化学图论中,化合物结构通常表示为氢贫化(省略氢)的分子图,每个化合物都以无向图表示,原子为节点,键为边。原子和键均包含很多属性例如原子类型、键类型等。文献[7]利用节点(原子)和边(键)的属性建立图卷积模型。文献[8]创建了原子特征向量和键特征向量,并将二者拼接形成原子键特征向量。文献[9]提出了图记忆网络(graphMem),这是一种记忆增强的神经网络,该网络可用于处理具有多种键类型的分子图。MPNN[10]阶段性地总结了GNN模型,摒弃手工特征,迈出了将GNN应用于分子图的重要一步。SchNet[11]推动了GNN在分子动力学模拟中的应用,使之符合物理学约束方程。DimeNet[12]对分子中的方向性信息进行建模,使得模型的预测精度更进一步。在这些研究中,都未对节点特征和键属性加以区分,没有关注其内部联系。但事实上,为原子对之间的各种相互作用类型赋予不同权重才是较为准确的方法。
最近,文献[13]提出一种基于边注意的图卷积神经网络算法(edge attention graph convolutional network, EAGCN),该算法提出了一个边缘注意层来评估分子中每条边的权重:预先构建了一个属性张量,经过注意层处理后,生成多个注意权重张量,其中每个张量都包含数据集中(分子图)一个边属性的所有可能的注意权重。然后,通过查找该权重张量中分子的每个键的值来构建注意力矩阵。这种方法使得模型可以在不同层次和不同边属性上学习不同的注意力权重。经实验证明,EAGCN框架具有很高的适用性,并且直接从图结构中学习特定的分子特征,避免了数据预处理阶段带来的误差。
本文基于EAGCN框架,考虑到无法自适应学习特征重要度带来的不稳定性,提出了基于多特性融合的注意力图卷积模型(multi-feature fusion dge attention graph convolutional network, MF_EAGCN),其中的多特性融合方案是基于自注意力机制的特征融合方式,能够有效地让模型自适应调节多个特征张量的权重分配。本文使用多种筛选方法对PubChem数据库中的靶标等内容作出限制,选择了不同类型的几种生物活性数据集,并将本文算法与几种基准模型同时应用于其中,分析评估了各自的性能。
-
在化学图论中,化合物结构通常表示为氢贫化的分子图,每个化合物以无向图表示,原子为节点,键为边。其中,分子的属性信息包括原子属性和键属性[14],具体描述见表1和表2。这些属性对于描述两个原子之间的键合强度、芳香性或键合共振等特征非常重要。如果将不同的边属性进行注意层处理,则不同的边属性对应于不同的边注意矩阵。
原子属性 描述 值类型 原子序号 原子在元素周期表中的位置 Int 相连的原子个数 邻居节点的个数 Int 相邻氢原子个数 氢原子数量 Int 芳香性 是否具有芳香性 Boolean 形式电荷个数 形式电荷个数 Int 环状态 是否在环内 Boolean 键属性 描述 值类型 原子对类型 键连接的原子类型定义 Int 键序 单键/双键/三键/芳香键 Int 芳香性 是否具有芳香性 Int 共轭性 是否共轭 Boolean 环状态 是否在环内 Boolean 占位符 原子之间是否存在键 Boolean -
定义1 图使用
$ G = \left( {V,E} \right) $ 表示,V为节点的有限集,$ \left| V \right| = N $ ,N为节点数,$ E \subseteq V \times V $ 是边的有限集合。定义2 G的邻接矩阵A是一个方阵,维度为
$ N \times N $ 。$ {a_{ij}} = 1 $ 代表节点i和j之间有连边,反之$ {a_{ij}} = 0 $ 则代表节点间无连边。定义3 为G构建一个节点特征张量
$ {H^l} \in $ $ {R^N} \times {R^F} $ ,F为每个节点的特征总数。第i行表示节点i的特征和一系列边属性,这里令K为边属性个数。定义4 假设对于边属性i,有
$ {d}_{i} $ 种可能的类型。定义5 为G构造一个分子属性张量
$M \in $ $ {R^{{N_{{\text{atom}}}} \times {N_{{\text{atom}}}} \times {N_{{\text{features}}}}}}$ ($ {N_{{\text{features}}}} $ 即为定义3中的F)作为注意层的输入。 -
EAGCN[13]在不同层次和不同边属性上学习不同的注意力权重,从而构建一个分子的注意力矩阵。该算法预先构建了一个属性张量,经过注意层处理后,生成多个注意权重张量,其中每个都包含数据集中一个边属性的所有可能的注意权重。然后,通过查找该权重张量中分子的对应键值来构建注意力矩阵。这种方法使得不同分子可对应不同的注意力矩阵。
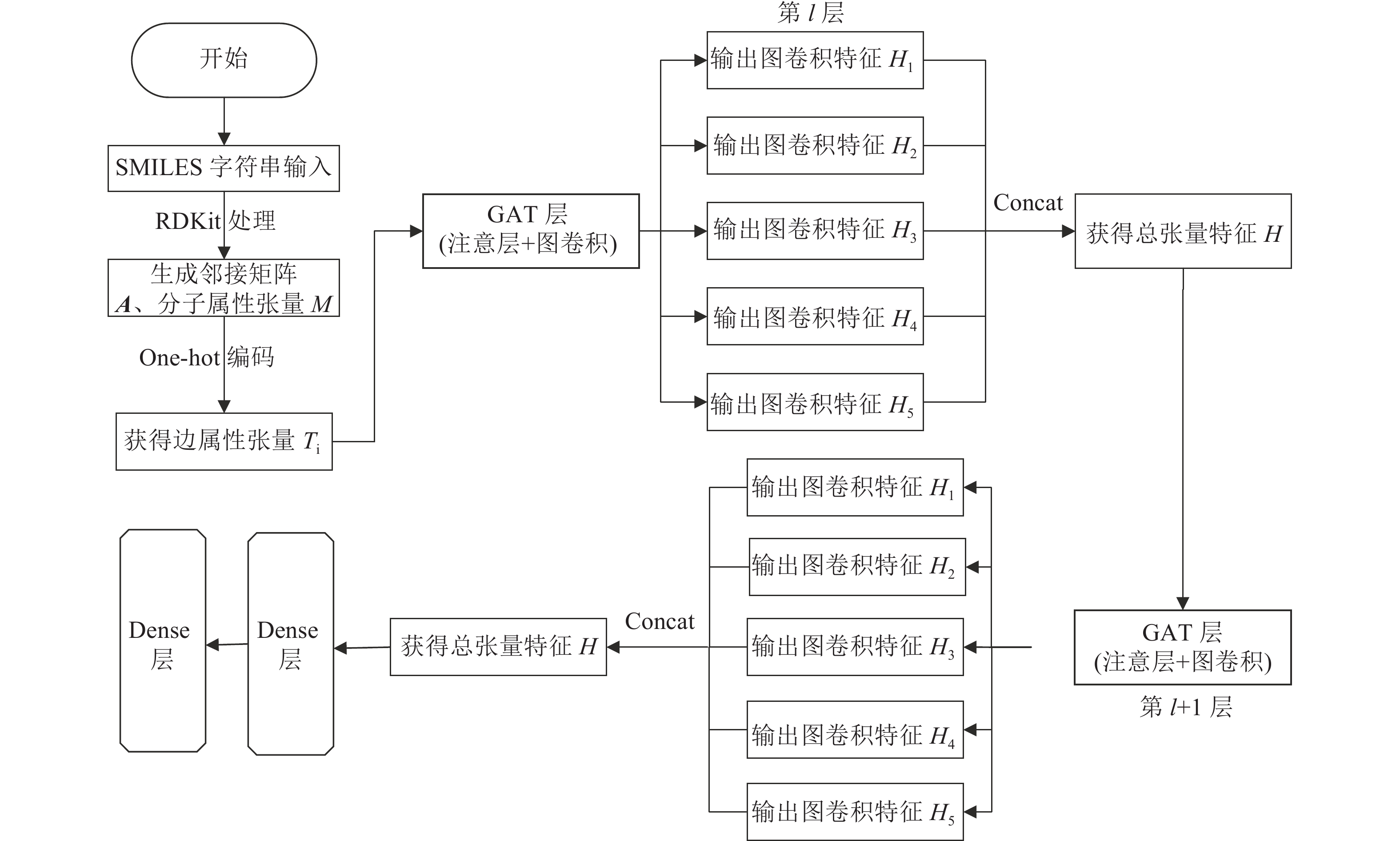
EAGCN利用分子的原子和键属性,为每个分子构建1个邻接矩阵A、1个节点特征张量
$ {H^l} $ 和1个分子属性张量M用于模型训练。模型总流程如图1所示,整个模型将分子图作为输入,处理分子图中的边属性后得到边属性张量,one-hot编码后分别经过GAT层得到5个图卷积特征,再经过concat拼接方式获得总张量特征,以此作为下一层GAT层的输入。最后使用两层dense层输出结果。
1) 注意层——
${{\boldsymbol{A}}_{{\rm{att}},i}}$ (图的注意力矩阵)构建过程上文介绍了本算法中的分子图属性特征选择,由以上可得邻接矩阵A和分子属性张量M。为计算得到每种属性中不同边的权重,将分子属性张量M按K条边属性拆分(本文选用表2中的前5个边属性,即K=5)。由于每个边属性含有不同的状态值,因此one-hot编码,会生成5个维度为
$ N \times N \times {d_i} $ 的邻接张量$ T_i^l \in {R^{N \times N \times {d_i}}} $ ,$ {d_i} $ 为边属性类型数。然后,通过边属性张量$ {T}_{i} $ 获得$A_{{\rm{att}},i}^l$ 中的权重$ a_{i,j}^l $ :这里,
${T}_{i}^{l}\to {A}_{{\rm{att}},i}^{l}$ 的处理过程为:先通过具有
$ {d_i} $ 个输入通道和1个输出通道的卷积处理,使用尺寸为$ 1 \times 1 \times {d_i} $ 的过滤器$ {D}_{i}^{l} $ ,以1为步长移动。其中,l表示在第l层边注意层。其次为了使权重在不同边中具有可比性,使用softmax函数对权重进行归一化,如式(2)所示。softmax函数又称为归一化指数函数,得到的输出值互相关联,它可以将其量化到0~1范围内,将多分类的结果以概率形式输出,且输出值总和为1。
简单来说,Attention层会通过二维卷积层(kernel_size为
$ 1 \times 1 $ )和softmax,对边属性张量$ {T}_{i}^{l} $ 进行操作,为每个边属性得出一个边权重张量${A}_{{\rm{att}},i}^{l}$ 。2) 图卷积层
在一个邻接矩阵中,图卷积只关注一个节点相邻节点的信息,即只取局部信息。在每个图卷积层中,对所有一阶邻居进行节点信息聚合,然后进行线性变换。通过式(3)计算l+1层的特征张量:
式中,i的范围为:1≤i≤K,K为边属性个数。σ为激活函数。每个边属性i会生成
${\tilde A_{{\rm{att}},i}}$ 里的一个值,因此${\tilde A_{{\rm{att}},i}}{H^l}$ 可以看作是节点特征的加权总和。接下来将不同边属性得到的特征张量$H_i^{l+1}$ 进行拼接,这里直接使用concat方式,最终形成总的特征张量$ {H^{l + 1}} = \left\{ {H_i^{l + 1} \in {R^N} \times {R^{{F_i}'}}|1 \leqslant i \leqslant K} \right\} $ 。重复步骤1),2),再进行
$ {H^{l + 1}} $ 至下一层的图卷积特征提取。经过两层GAT层处理后,最后再衔接两个全连接层进行分子模型的分类计算,得出分类置信度。 -
本文将EAGCN用于本文收集的不同种类的生物活性预测数据集,得到了比传统机器学习更好的模型性能。而EAGCN模型的某些特性是使得其在大多生物活性数据集上性能较优的原因:
1)其直接对分子图进行学习,可以很好地避免人工筛选特征带来的误差,一定程度上提升了模型的鲁棒性和可靠性;
2)其生成的注意权重矩阵取决于一个节点的领域特性,而不是全局特性;且权重可在所有图中共享,于是可通过共享的特征来实现数据的局部特性提取。
在原始模型中,权重张量经过图卷积处理得到特征后,整合特征图信息时常使用concat方式合并通道。concat经常用于将特征联合、多个算法框架提取的图特征融合又或是将输出层的信息进行融合,将融合后的特征作为下一个网络层的输入。concat虽然较为常用,但也存在一些问题:其只是简单的特征张量的维度拼接,相当于只是通道数的增加。这只是增加了图像本身的特征,对于多特征的重要度分析并没有起到太大作用。这不仅会导致多个属性信息没有区分度,增加维度还可能会降低模型的计算效率,影响模型性能。于是本文提出使用多特性融合的方式替换concat方法。在EAGCN中,注意力机制被用于从邻居节点那里学习节点对之间边的交互强度,简单来说是为了得知边在整个图中的重要性。经过实验可知“原子对类型”这一边属性对整个模型性能影响较大,因此在设置网络通道数参数时,本文将为原子对类型的特征矩阵设置更高的通道数,相当于使用人工设置偏向权重的方法,这种方法存在一定的不稳定性。
为了更科学地知道每种边属性特征的重要性,且能够有效地让模型自适应调节多个特征张量的权重分配,本文提出了多特性融合的方法进行算法优化。这是基于自注意力机制(self-attention)[15]的特征融合方案,它可以对输入的每个元素赋予不同的权重参数,从而“挑出”每种特征中较为重要的信息,抑制但不丢失其他信息。其最大的优势就是能一步到位地考虑全局联系和局部联系,可以进一步提高模型的学习效率。
EAGCN为每张图生成了分子属性张量
$M$ ,为了计算得到每种属性中不同边的权重,将分子属性张量M进行one-hot编码,再将多个属性张量输入注意层,进而得到多个边权重张量${A^l}_{{\rm{att}},i}$ 。经过图卷积层的处理后得到特征张量$ {H^{l + 1}} $ ,将concat融合方式替换为多特性融合方案,具体步骤如下。1)为每个输入生成Q、K、V张量
将得到的5个特征张量
$H_i^{l+1} $ 作为输入。$ {H_i^{l + 1}} $ 的维度根据模型中设置的通道数而变化。以一个维度为N×30的图特征张量$ H_i^{l+1}$ 为例,先为每个特征张量设置3个不同的张量,分别为查询Q、键K、值V,长度默认为64。$ {W^Q},{W^K},{W^V} $ 是3个不同的权重张量(3个张量维度相同,都为30×64),用特征张量$ {H_i}^{l + 1} $ 分别与它们相乘,得到对应的Q、K、V张量,计算示例如图2所示。上述过程在计算时其实是基于矩阵运算的,即运算时是将输入张量合并计算的。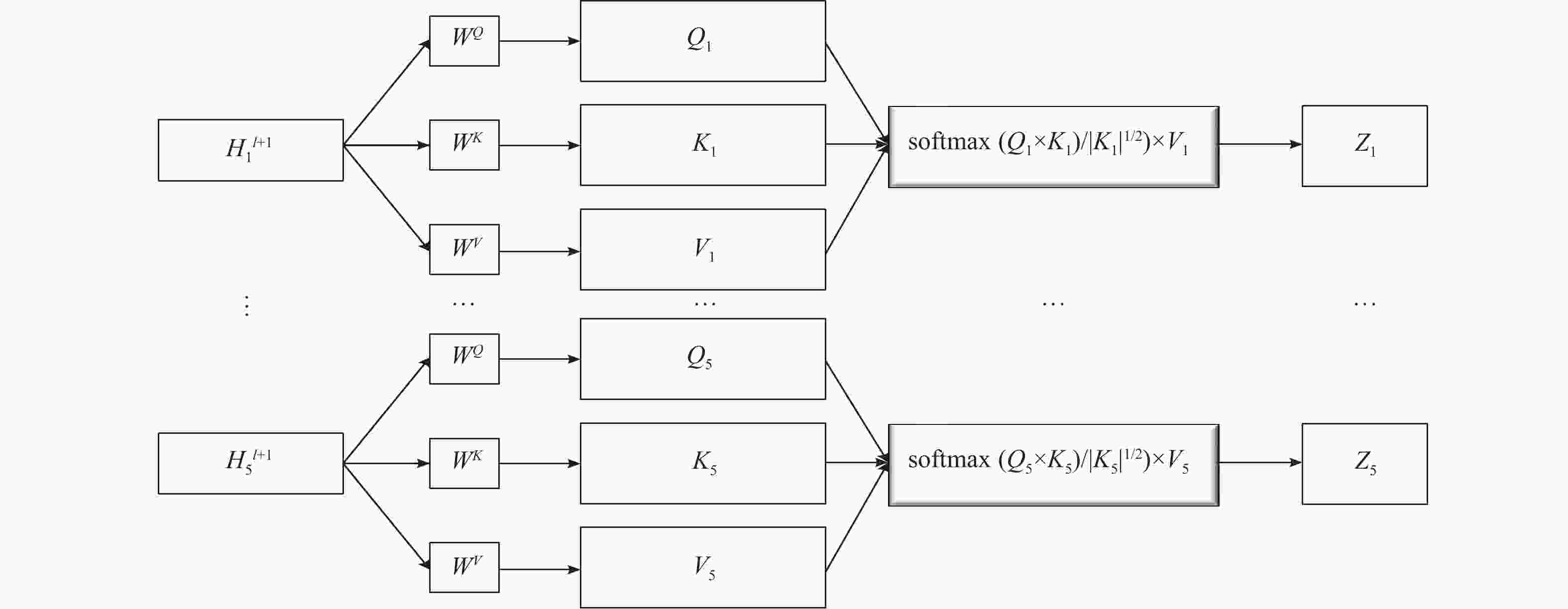
2)计算得分
将每个特征的键向量和查询向量进行点积运算,得到其分数:
${\text{score}} = Q \times K$ 3) score归一化
为了让梯度更稳定,将步骤2)中计算得到的score除以
$ \sqrt {{d_k}} $ 进行归一化。$ {d_k} $ 为键张量K的模长,即64。4) softmax归一化
使用softmax对所有特征张量的score进行归一化,使得到的score都为正且和为1。这一步的目的是初步得到每个边属性对于整个图的权重。
5)求输出张量评分
将值张量V与softmax分数点乘,得到加权的每个输入张量(图卷积特征)的评分v。
6)对加权值向量求和
得到输出:
$z = \displaystyle\sum v$ ,$z \in {R^N} \times {R^{30}}$ ,即为权重矩阵z。 -
本文所选用的数据集来自于一个公共化学数据库PubChem[16]。本文选用了文献中的多种分析筛选方法[14],选择了相同类型和不同类型的生物活性数据集,对筛选的靶标等作出了限制,如筛选了细胞色素P450酶的多个系列。最终本文选用了1851靶标家族中细胞色素酶P450系列的4个数据集、两种抑制剂和识别结合r(CAG) RNA重复序列的分子系列。表3列出了所选用的数据集的相关信息以及筛选条件。
PubChem AID 筛选条件 有活性分子数 无活性分子数 1851(1a2) Cytochrome P450, family 1, subfamily A, polypeptide 2 5997 7242 1851(2c19) Cytochrome P450, family 2, subfamily C, polypeptide 19 5905 7522 1851(2d6) Cytochrome P450, family 2, subfamily D, polypeptide 6,isoform 2 2769 11127 1851(3a4) Cytochrome P450, family 3, subfamily A, polypeptide 4 5265 7732 492992 Identy inhibitors of the two-pore domain potassium channel (KCNK9) 2097 2820 651739 Inihibition of Trypanosoma cruzi 4051 1326 652065 Identify molecules that bind r(CAG) RNA repeats 2969 1288 -
分子的生物活性研究中,输入数据是QSAR研究的基础,不同的算法模型所使用的分子输入数据形式也是不同的。分子的表示形式常见的有:分子标识符、分子描述符两种。
分子标识符是基于文本的标识符,如简化分子线性输入规范(simplified molecular input line entry system, SMILES)[17]和国际化学标识符(InChl)[18]。SMILES是用一组有序规则和专门语法将三维化学结构编码的文本字符串[17],是一种用于存储化学信息的语言结构。如二氧化碳(CO2)的SMILES标识符为O=C=O。SMILES是目前QSAR建模中较常使用的标识符。
国际化学标识符InChl用不同的化学信息层(连通性、立体化学、同位素和互变异构体)来表达化学结构[18]。但后期多项研究发现,其复杂的数字公式会导致预测性能下降,因此并未在深度学习中经常使用。
分子描述符是早期QSAR研究的基础,传统机器学习模型无法识别及处理分子结构,将分子的物理化学性质或分子结构相关参数,利用各种算法推导出模型可以处理的数值。
目前,用于分子描述符的计算工具有很多种,包括各种开源或商业软件及各种开源库。可以生成的分子描述符已接近10000个,包括1D、2D、3D描述符以及一些指纹描述符等。近些年,常用的分子描述符计算软件有Dragon[19]、alvaDesc[20]、Gaussian[21]、Padel-Descriptor[22]、OpenBabel[23]等。其中,经典的Dragon软件已迭代到7.0版本,可以计算几千种分子描述符,但不幸的是已经停产,进而代替它的是alvaDesc。alvaDesc可计算5305种分子描述符(包括Dragon 7中可用的所有描述符),以及一些特殊描述符如MACCS指纹的计算。常用的化学库有RDkit[24]等。RDkit是非常著名的开源化学信息软件包,提供了Python和C++语言的API接口,不仅可以计算各种分子描述符,还可以进行分子可视化及化学分析等工作,适用性极好。
本文实验将MF_EAGCN与EAGCN、随机森林(random forest, RF)、支持向量机(support vector machines, SVM)及深度神经网络(deep neural networks, DNN)用于相同的数据中。在传统机器学习方法中(RF、SVM、DNN),需要使用计算生成的分子描述符,因此本文在设计实验前,对于分子SMILES数据,使用RDKit(开源化学计算软件包)生成的200个一维分子描述符作为基准模型的特征;同时将RDKit计算出的分子的原子属性、边属性用于本文算法。
-
首先将EAGCN应用于本文选用的不同类型生物活性分类数据集,然后将基于多特性融合的注意力图卷积应用于同样的数据集中。本节设计实验的目的是:1)验证基于边注意的图卷积模型相较于传统机器学习方法(如随机森林、深度神经网络等)确实更能提升对生物活性数据的分类性能,且由于数据的多样性,模型在生物活性预测问题中也具有一定的普适性;2)验证本文针对特征融合方式进行优化得到的模型——基于多特性融合的注意力图卷积模型,在生物活性预测任务中的性能提升。
为了降低传统留一法划分数据法中的偶然性,提高泛化能力,使得数据使用率高,且考虑到算法复杂度,模型的数据集划分选用K折交叉验证方法,设置K=8,然后用不同的随机种子执行3次。同样,这里得到的结果均为3次运行的平均值,并列出了标准偏差。
-
本文使用的基准方法为RF、SVM及DNN 3种。针对3种模型,如表4所示,设置了超参数列表进行模型调参。同样的,数据集划分选用八折交叉验证法,然后用不同的随机种子执行3次。这里得到的结果均为3次运行的平均值,并列出了标准偏差。
-
在EAGCN建模时根据分析得到,原子对类型这一属性的权重设置较大时,模型性能会较好,于是在该算法中人工将原子对类型的GCN层输出通道数设置的偏大,为了更好地学习此特征,做出了人工干涉。在优化的MF_EAGCN中,会自行关注较高权重的边属性,即可以自适应的学习不同的边属性权重。本文设置的实验参数如表5所示。
基准方法 超参数 值区间 参数意义 RF Ntrees ($50,100,\cdots ,500$) 树的个数 max_depth ($1, 5,\cdots ,50$) 每棵树最大树深度 max_features $(1, 5,\cdots ,50$) 划分时的最大特征数 SVM Kernel RBF 核函数 C (1,10,100) 惩罚系数 γ (0.1,0.001,0.0001,0.00001,1,10,100) 影响数据映射到新特征空间的量 DNN Epoch 100 迭代次数 Batch size 100 最小训练样本数 Hidden layers (2,3,4) 隐层数 Number neurons (10,50,100,500,700,1000) 每层神经元个数 Activation function ReLu 神经元激活函数 Loss function binary_crossentropy 损失函数 模型 超参数 值区间 参数意义 EAGCN Batch size 64 单次训练样本数 Epoch 100 迭代次数 weight_decay 0.00001 权重衰减率 dropout 0.5 随机失活率 Activation function ReLu 激活函数 Loss function binary_crossentropy 损失函数 kernel_size 1 卷积核大小 stride 1 卷积核滑动步长 n_sgcn1 (30, 10, 10, 10, 10) 多特征图卷积层输出通道数 MF_EAGCN Batch size 64 单次训练样本数 Epoch 100 迭代次数 weight_decay 0.00001 权重衰减率 dropout 0.5 随机失活率 Activation function ReLu 激活函数 Loss function binary_crossentropy 损失函数 kernel_size 1 卷积核大小 stride 1 卷积核滑动步长 n_sgcn1 (20, 20, 20, 20, 20) 多特征图卷积层输出通道数 -
本文使用两种评价指标:准确率(accuracy, ACC)和平衡F1分数(balancedscore, F1-score)。
其中准确率(ACC)是分类预测中较为常用的评价指标:
式中,TP、TN分别为被正确地划分为正例、负例的个数;P、N为实际样本中正例、负例的个数。总的来说,ACC就是被分对的样本数占所有的样本数的比例,ACC指标值越高,分类器性能越好。
平衡F分数F1-score也是生物活性分类任务中常用来衡量模型精确度的指标:
F1-score同时考虑到了模型的精确率(precision)和召回率(recall),只有在两个值都高时,F1的值才会更高,模型性能越好。其中,precision与recall的计算公式如下:
式中,FP、FN分别表示被错误的划分为正例、负例的个数。
-
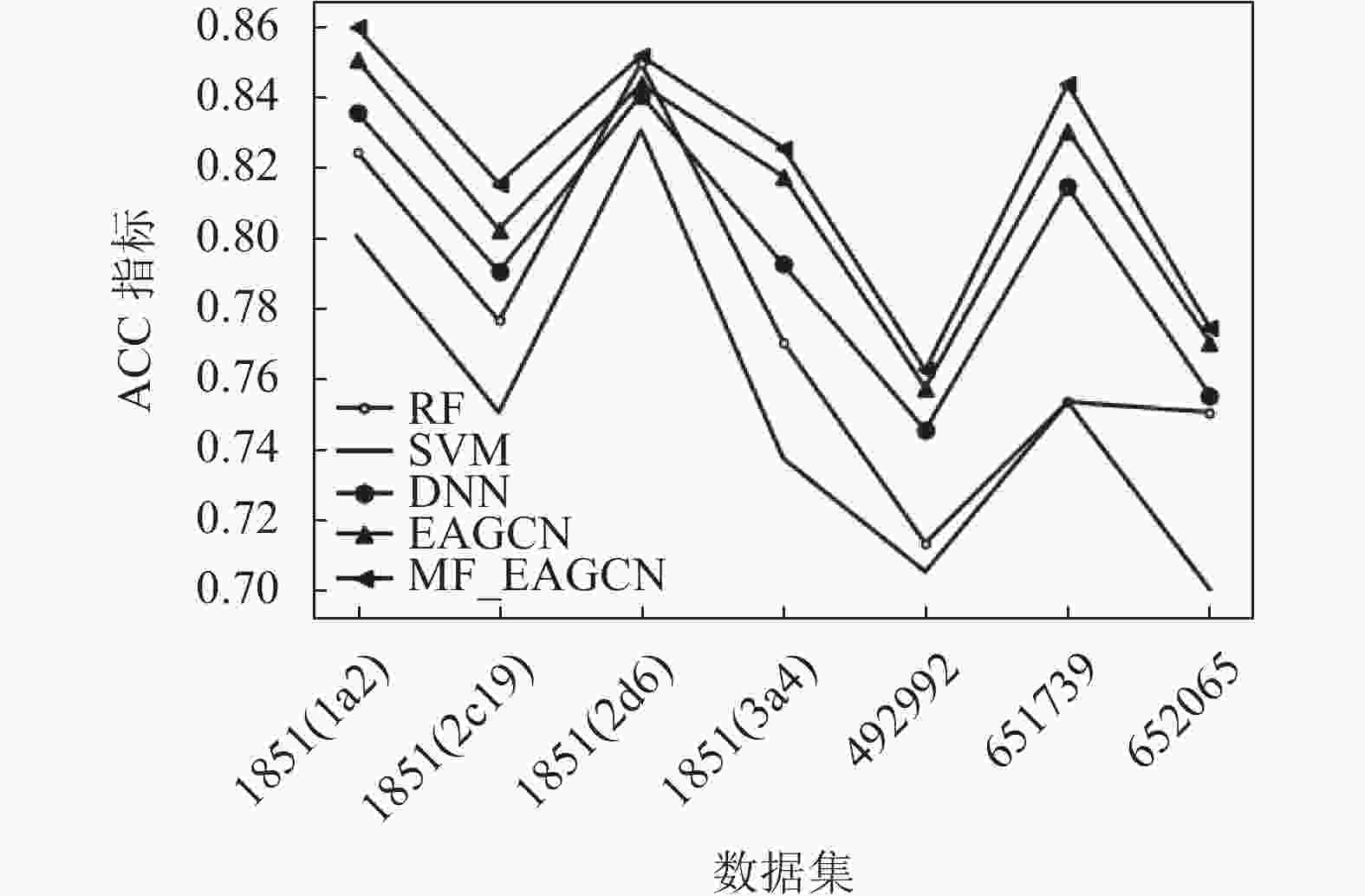
表6显示了在几种数据集上,不同基准模型的ACC、F1-score指标结果。
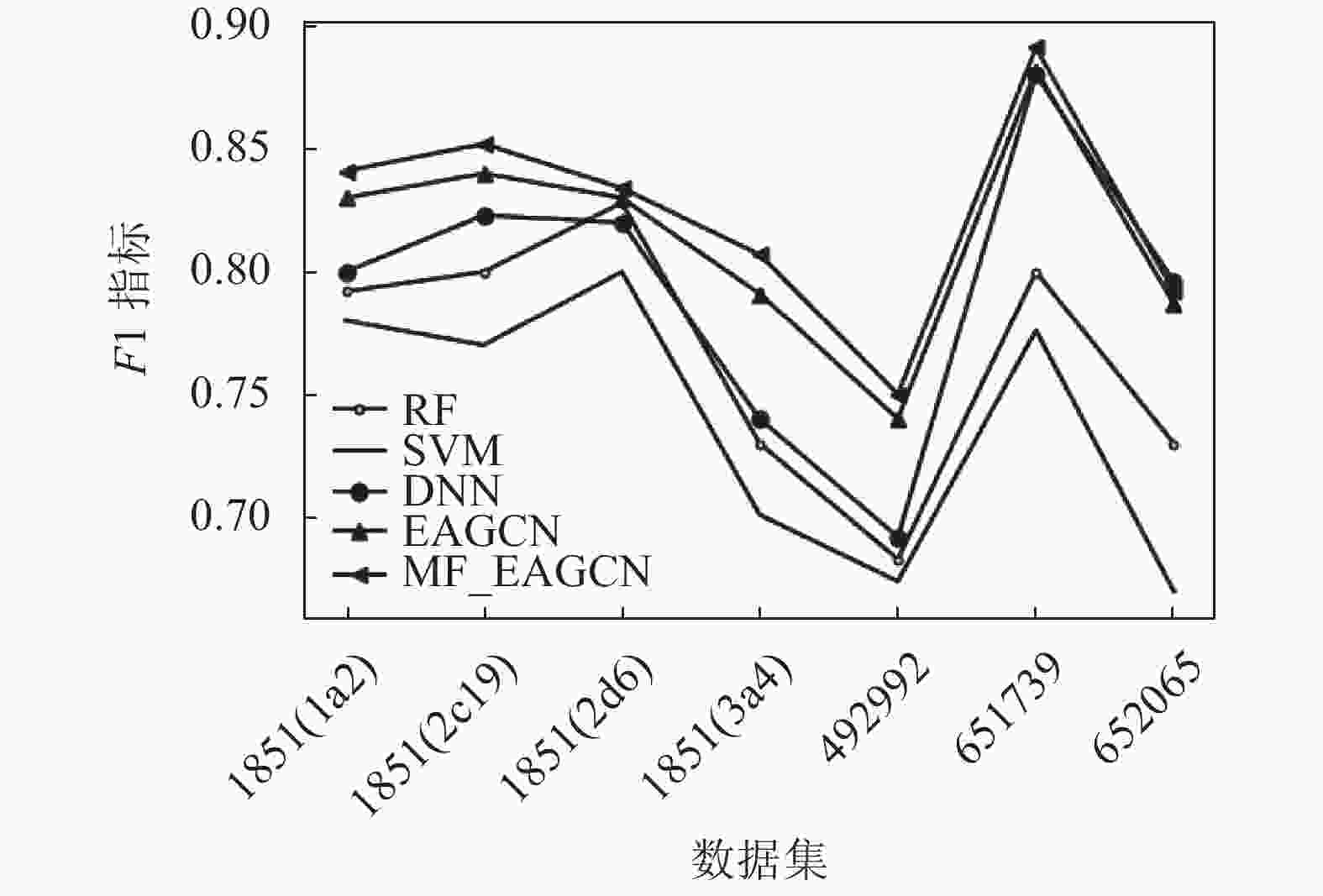
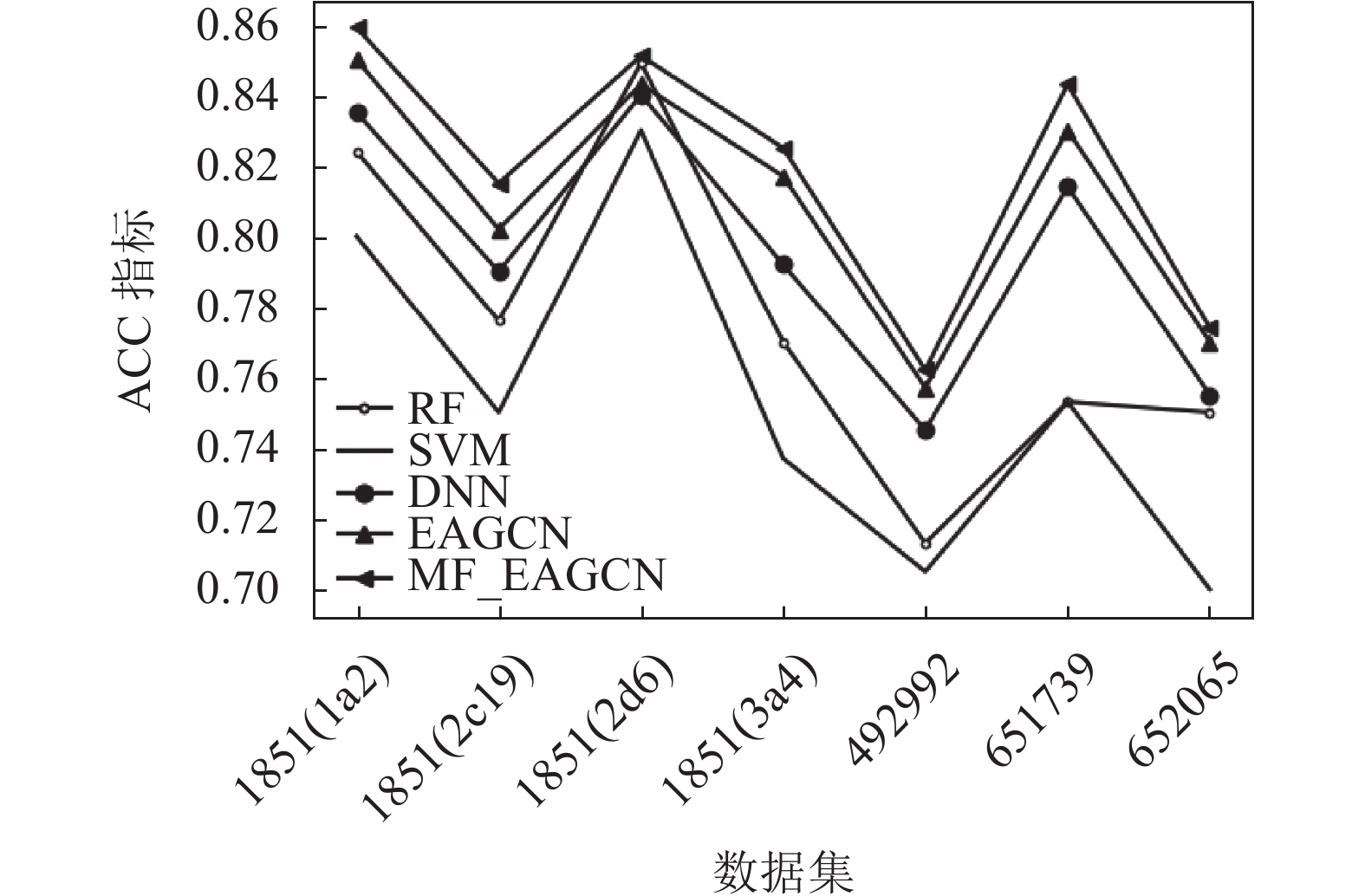
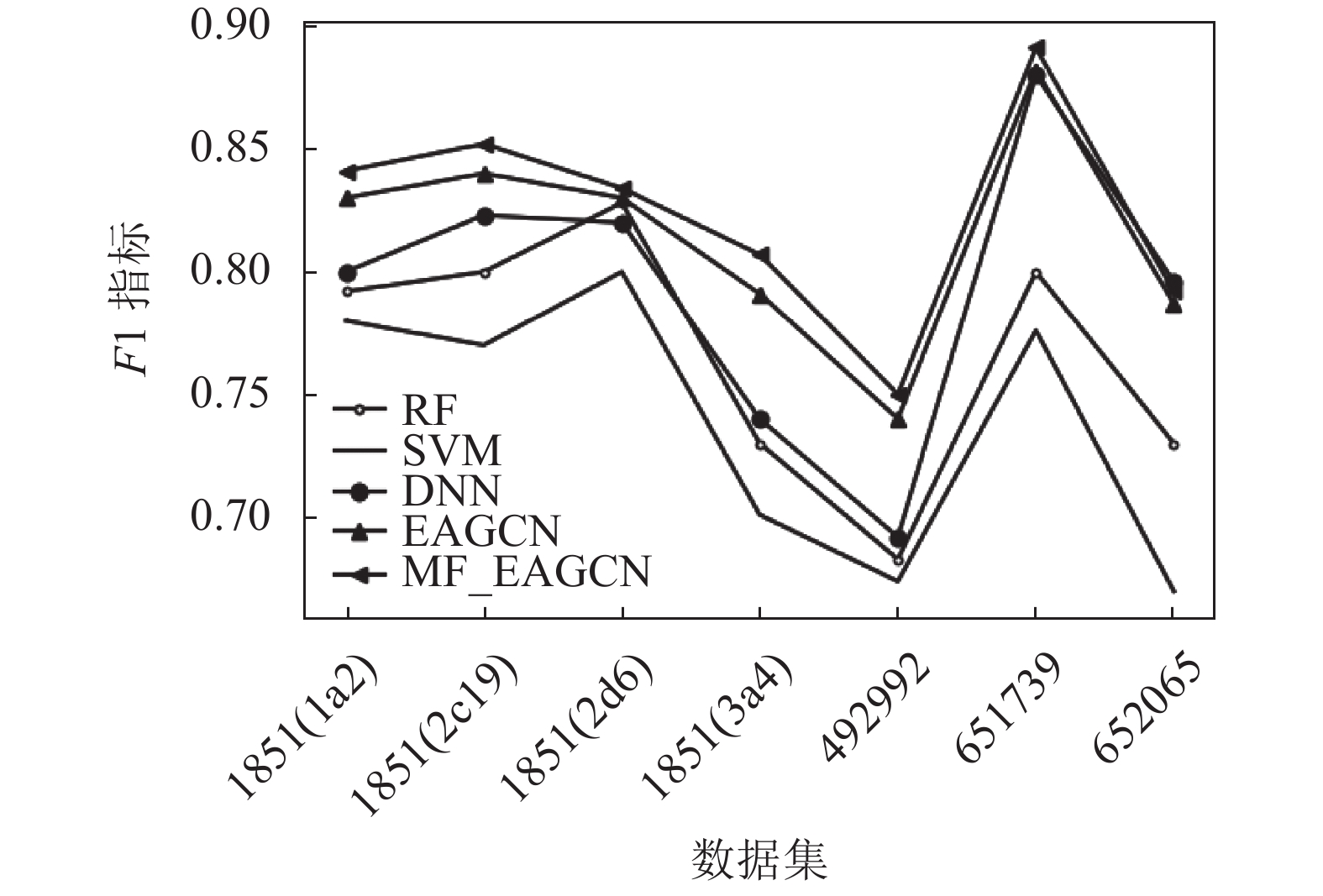
从实验结果可以看出,在这些数据集中,基于图卷积的EAGCN展现出了比传统机器学习方法更好的分类性能,其ACC指标均比基准学习模型高出2%~8%,F1-score指标比基准学习模型高出1%~5%。可见直接从分子图学习而不是从预先计算的特性中获得的信息使得模型性能更优。少部分数据集中,DNN的性能能与EAGCN方法性能基本持平或稍微高于其性能,RF的性能有时可以与EAGCN持平。可见,EAGCN的性能还有很多优化空间。而基于多特性融合的MF_EAGCN模型,展现出了更好的分类性能,这也证实了多特性融合方案能够更充分地利用边属性信息进行特征提取,使得模型预测性能提升。其ACC指标均比EAGCN算法高出1%~2%,F1-score指标比EAGCN模型高出约1%。
数据集 ACC F1-score RF SVM DNN EAGCN MF_EAGCN RF SVM DNN EAGCN MF_EAGCN 1851(1a2) 0.824
±0.0050.800
±0.0200.835
±0.0150.850
±0.0100.859
±0.0120.792
±0.0100.780
±0.0080.800
±0.0070.830
±0.0120.841
±0.0101851(2c19) 0.776
±0.0100.750
±0.0090.790
±0.0020.802
±0.0070.815
±0.0030.800
±0.0040.770
±0.0050.823
±0.0100.840
±0.0100.852
±0.0081851(2d6) 0.849
±0.0060.830
±0.0070.840
±0.0020.843
±0.0050.851
±0.0030.828
±0.0130.800
±0.0040.820
±0.0030.830
±0.0100.834
±0.0061851(3a4) 0.770
±0.0060.737
±0.0040.792
±0.0080.817
±0.0060.825
±0.0100.730
±0.0030.701
±0.0060.740
±0.0100.791
±0.0080.807
±0.005492992 0.713
±0.0040.705
±0.0060.745
±0.0050.757
±0.0100.762
±0.0100.683
±0.0050.674
±0.0060.692
±0.0090.740
±0.0100.750
±0.009651739 0.753
±0.0040.753
±0.0060.814
±0.0140.830
±0.0060.843
±0.0030.800
±0.0030.776
±0.0090.880
±0.0060.882
±0.0070.891
±0.002652065 0.750
±0.0040.700
±0.0050.755
±0.0150.770
±0.0060.774
±0.0050.730
±0.0080.670
±0.0090.796
±0.0120.787
±0.0100.792
±0.010图3和图4分别展示了本文提出的MF_EAGCN、基准算法EAGCN以及传统机器学习方法5种分类器,分别应用于7种生物活性数据集中的ACC指标和F1-score指标分布对比,柱状图的条目从左到右依次是RF、SVM、DNN、EAGCN和MF_EAGCN模型。在ACC指标分布图中,可以看到数据集1851(2d6)在EAGCN模型上的效果并不显著,其原因可能是由于数据量相比较而言更大,在模型融合特征阶段对特征重要度分配不均,导致对重要信息的忽略,进而致使模型预测性能降低。而本文提出的MF_EAGCN模型很好地缓解了此问题,相较于EAGCN,其预测性能提升了2个百分点,而相较于基准机器学习模型,其预测性能提升了8个百分点,由此也验证了本文算法的有效性。
-
本文提出了基于自注意力机制的多特性融合方案,针对基于边注意机制的图卷积网络模型进行了有效优化。本文将一种基于边注意力的图卷积网络架构,应用于文中选用的不同种类的生物活性预测任务,从而避免了人工特征工程带来的误差,并对比几种机器学习基准算法,验证了本人算法有效性。在此基础上,针对前人提出的模型中存在的问题:无法自适应设置边属性特征权重,本文提出了分子多特性融合的方案优化了算法模型的特征提取能力,通过自注意力机制针对多个特征进行自适应融合,有效地解决了这一问题,并且获得了更好的预测性能。本文使用的数据集偏向数据量较小的数据集,未来会将其扩展到数据量更大的数据集以及其他生物活性预测任务上。在应用于较大数据集时,模型可以针对性地对不同任务作出优化,可以提高模型的泛化性能,提升模型稳定性。
Prediction of Molecular Biological Activity Based on Graph Convolution Method of Multi-Characteristic Fusion
doi: 10.12178/1001-0548.2021158
- Received Date: 2021-06-21
- Rev Recd Date: 2021-10-02
- Available Online: 2021-11-26
- Publish Date: 2021-11-28
-
Key words:
- graph convolution /
- multi-characteristic fusion /
- peripheral attention /
- prediction of molecular biological activity
Abstract: The development cycle of drugs is long and the cost is huge. The method of computerized virtual drug screening can effectively improve the efficiency of the pilot compounds. This paper proposes a new feature fusion scheme based on attention mechanism, called multi-feature fusion scheme. Combined with the existing graph convolution network based on edge attention, the biological activity prediction task is carried out by using this method for different kinds of bioactive data sets selected from PubChem, the public chemical database. The instability and unreliability caused by manual calculation can be avoided by learning the molecular graph features directly, and multi-feature fusion scheme based on attention makes the model adaptive to fuse multiple edge attribute features. The results show that the method can predict the biological activity of molecules more accurately than other machine learning methods.
| Citation: | TAN Lulu, ZHANG Xinxin, ZHOU Yinzuo. Prediction of Molecular Biological Activity Based on Graph Convolution Method of Multi-Characteristic Fusion[J]. Journal of University of Electronic Science and Technology of China, 2021, 50(6): 921-929. doi: 10.12178/1001-0548.2021158

|















































 DownLoad:
DownLoad: