 ISSN
ISSN 





-
随着人口的增长及老龄化,恶性肿瘤(即癌症)已经成为人类死亡主要原因之一,是威胁生命健康的最大因素[1]。肿瘤癌变是环境因素和遗传因素引起基因突变造成的,识别癌症的致病基因对于精确肿瘤学至关重要,并且能够促进靶向药物的开发,对癌症的治疗具有指导意义[2-4]。
随着第二代测序技术普及,以及人类基因组计划、TCGA计划和ICGC计划的推进,研究者从大规模测序分析结果中明确了肿瘤存在着广泛的异质性[5]。分布在不同患者个体中或者同一患者体内不同部位中的同种恶性肿瘤细胞,会产生从基因型到表型上的差异,相应地表现为多样的基因突变[6]。高通量的测序数据使得科学家能从蛋白质水平中揭示肿瘤细胞发生机制,验证肿瘤的相关基因突变,在癌症中的应用有着广泛的前景。文献[7]证明了乳腺癌的蛋白质组学分析能够解读体细胞突变,缩小了缺失和扩增区域内驱动基因的候选提名范围,并发现了相关治疗靶标。文献[8]从蛋白质组学研究入手,发现了白血病抑制因子(Leukemia inhibitory factor, LIF)是介导胰腺癌细胞和星状细胞之间信号传导的关键因子,并验证了其可以作为胰腺癌治疗的靶点和生物标志物。
然而,面对庞大的肿瘤数据,精准地找到肿瘤关键基因仍面临着挑战,仅从生物学角度研究肿瘤关键基因是远远不够的。如今,癌症这个复杂疾病又被称为“网络疾病”,可以从网络的角度对生物学进行研究[9-10]。网络中的节点可以代表生物分子,其相应的边可以看作生物分子之间的功能、物理或化学相互作用[10]。因此,挖掘肿瘤的致病关键基因,找到潜在的控制疾病进展的靶点,可以从网络控制入手。复杂网络的控制理论和方法源于经典控制理论与复杂系统研究的结合,如果网络中的一部分节点能够在有限时间内将网络从任意初始状态变为任意期望的最终状态,则该网络称为可控网络[10- 11]。文献[12]在复杂网络可控性方面做出了开拓性的研究,将网络的可控性简化为判定网络结构可控性的问题,即忽略网络中系统矩阵的边权,只需关注系统内部的结构框架及节点间的连接方式。然而,文献[12]提出的最大匹配算法只适合在有向网络中寻找控制节点。为了应用于无向网络,文献[13]提出了最小控制集(minimum dominating set, MDS)方法,并发现MDS方法在无标度网络中只需要较小比例的节点就可以覆盖控制网络,并且网络度分布的异质性越强,就越容易控制整个系统。
如今,复杂网络控制理论已被广泛应用到各种生物网络分析中。文献[14]在有向人类蛋白质相互作用网络中应用最大匹配算法[12]确定了最小驱动蛋白集合,根据移除驱动节点后所包含的驱动节点数,对基因进行分类。此外,同时发现该可控性分析在疾病基因以及药物靶点上的有关键作用。文献[15]利用MDS方法研究了蛋白质相互作用网络的可控性,并确定了驱动蛋白集,分析表明该集合富含必需基因、癌症相关基因以及病毒靶向基因,并且这些集合在网络调控(富含转录因子和蛋白激酶)中有着重要作用。文献[16]在MDS模型的基础上提出了一种中心性校正的最小支配集(centrality corrected-MDS, CC-MDS)模型,该模型比MDS模型能够捕获更多的驱动蛋白。文献[17]利用网络的无标度特性算法[18]巧妙地避开了MDS计算的复杂性,利用MDS各个解集中元素的角色,将节点分为3类并应用于蛋白质相互作用网络中,首次捕捉到了基因结构可控性、基因致死性和动态共表达的同步性之间的直接联系。虽然网络控制的方法可以捕捉生物网络中的关键驱动蛋白及重要的相关调控功能,但是其对于各类癌症中肿瘤关键基因识别预测方面还未得到分析验证。
本研究利用复杂网络控制理论的思想,从蛋白质相互作用网络(protein protein interaction network, PPI)入手,通过对肿瘤发生的不同阶段PPI的变化进行分析,对肿瘤关键基因进行预测研究,可以阐明肿瘤蛋白表达水平的变化与肿瘤发生发展的相互关系及其规律。检测、分析和确定肿瘤不同时期的标志蛋白,可以作为抗癌药物筛选的作用靶点。该应用不仅对抗癌药物发现具有指导意义,还可形成未来肿瘤诊断学、治疗学的基础理论。
-
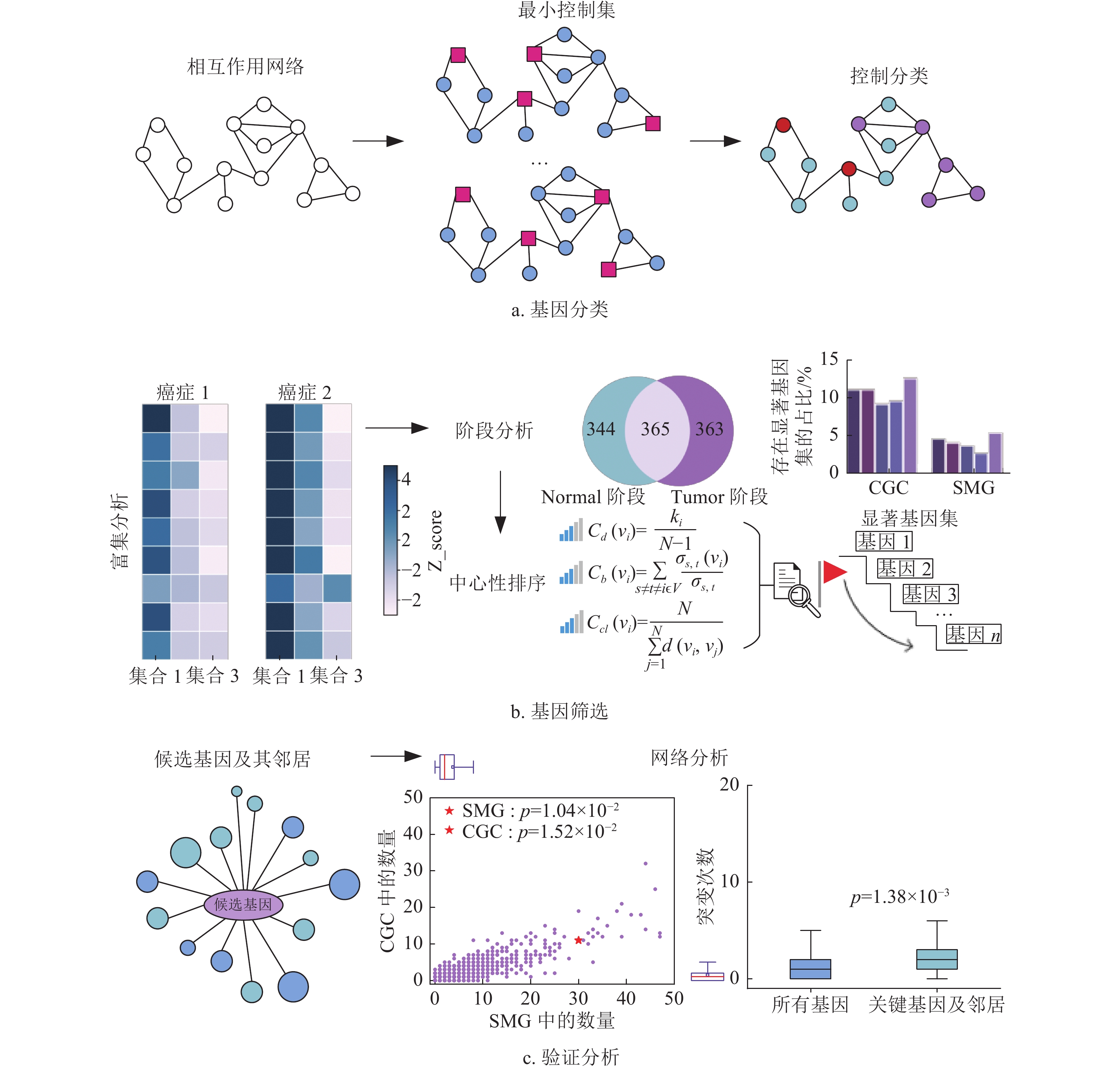
为了预测对肿瘤发展具有关键影响的基因,本文将复杂网络控制理论应用于人类相互作用蛋白数据中,结合控制集和复杂网络的拓扑性质,对蛋白质相互作用网络中的基因进行了筛选,并对最终预测的肿瘤关键基因进行了有效的验证。图1描述了肿瘤关键基因预测分析的流程,主要包含3个步骤:1)通过最小控制集和控制分类模型将蛋白质网络中的基因进行分类;2)对得到的各分类中的基因集合利用综合中心性进行排序,进行富集分析,筛选出最终可用来预测的肿瘤关键基因;3)将得到的肿瘤关键基因,通过突变数据及相应的文献分析,证明预测的肿瘤关键基因对肿瘤发展的影响力。Z_score衡量的是观测值与平均值间的差异,p指的是在假设检验中,当原假设为真时所得到的样本观察结果或更极端的结果出现的概率。
-
假设
$G = \left( {V,E} \right)$ 表示一个无向图,其中$V = $ $ \{ {v_1},{v_2}, \cdots ,{v_n}\} $ 为$G$ 的节点集,$N$ 为无向图中节点的个数,$E$ 为$G$ 的边集。如果从顶点$ {v_i} $ 到顶点$ {v_j} $ 中有路径存在,则称$ {v_i} $ 和$ {v_j} $ 连通。存在子图$G'$ ,如果其中的任意两个顶点之间都连通并且为连通的子图中最大的一个图,则该图$G'$ 为无向图$G$ 的最大连通子图。在蛋白质相互作用网络中,节点代表蛋白质,边代表两个蛋白质之间的相互作用关系。其最大连通子图就是蛋白质相互作用网络中的最大的一个连通网络,网络中任意两个蛋白质之间都能够直接(或间接)相连通。

图 1 肿瘤关键基因预测分析流程
-
图
$ G = (V,E) $ 中存在一个节点集$S \subseteq V$ ,如果节点${v_i} \in V$ ,其要么是节点集$S$ 的一部分,要么与节点集$S$ 里一个元素相邻,则称这样的节点集$S$ 为控制集(如图1a中间网络中的红色方形节点)。控制集$S$ 里的这些节点是图1a中的最优节点集,它对整个图的结构有着支撑作用,除此之外的每个节点都能够通过一条连边与控制集$S$ 里的一个节点相连。本文定义在图$G$ 的所有可能的控制集当中节点数最少的集合为MDS。MDS的概念属于图的控制理论。由于控制问题是经典的NP完全问题,要解决这个未知能否在多项式时间内求解的问题,需要把它归结为一个二进制的整数规划问题(integer linear programming, ILP)来计算。本文通过寻找二进制向量
$x$ 来最小化具有线性约束下的线性函数$f(x)$ 。图$ G = (V,E) $ 中每个节点${v_i}$ 的都有一个值为0或1的二进制整数变量${x_i}$ ,属于控制集的节点的二进制变量取值为1,否则为0。于是,线性函数$f(x)$ 的约束表示为:$$ f(x) = {x_1} + {x_2} + \cdots + {x_n} = \sum\limits_{i = 1}^n {{x_i}} $$ (1) 并且满足约束:
$$ {x_i} + \sum\limits_{j \in N(i)} {{x_j}} \geqslant 1 $$ (2) 式中,
$N(i)$ 表示节点${v_i}$ 的邻居;而$n$ 是图中的节点总数。最小控制集能够提供对整个网络起控制作用的节点集。蛋白质相互作用网络中的这些最小控制集,能够通过其相互作用影响整个PPI网络。解决最小控制集这个二进制整数规划问题,可以利用分支限界算法来寻找这二进制线性函数约束问题的最优解。
-
在一个连通的蛋白质相互作用网络中,寻找控制基因集的最优解,往往会有多种符合最优解的解决方案。单纯地将其中的一个最小控制基因集作用于复杂网络中,并不能确定始终能够影响整个网络的一些关键节点的角色(即对整个癌症网络有确定影响的基因集),因此需要对集合进行进一步的分类。为了解决这个问题,文献[13, 17]根据每个节点在不同配置中参与的角色,提出了分类算法,构建了控制分类模型:始终存在于配置方案的关键节点(critical)、从不参与任何配置方案的冗余节点(redundant)、属于某些配置方案但不存在于其他配置方案的间歇性节点(intermittent)。这3类节点分别对应着图1a右侧网络的红色、蓝色和紫色的圆形节点。为了优化计算速度,文献[13, 17]在分类之前利用所证实的推论,对网络中的节点进行了预处理,利用拓扑性质提前确定了一些节点的类型。
推论1 关键节点推论。如果节点
${v_i}$ 有两个或两个以上的度为1的邻居结点,那么这个节点${v_i}$ 被定义为一个关键节点。推论2 冗余节点推论。如果节点
${v_i}$ 所有的邻居结点都是关键节点,那么这个节点${v_i}$ 被定义为一个冗余节点。本文整合了该控制分类模型,并将网络中的节点进行了分类,算法流程如下所示。
算法1 节点的控制分类
输入:节点集
$V$ 、边集$E$ 输出:关键节点集
${G_{{\text{critical}}}}$ 、冗余节点集$ {G_{{\text{redundant}}}} $ 、间歇性节点集${G_{{\text{intermittent}}}}$ 1) 初始化
${G_{{\text{intermittent}}}}$ 2) for
$ {v_i} \in V $ do利用推论1得到关键节点集
${G_{{\text{critical}}}}$ 利用推论2得到冗余节点集
$ {G_{{\text{redundant}}}} $ end
3) for
${v_i} \in {G_{{\text{critical}}}}$ do通过式(1)将约束
${x_{{v_i}}} = 1$ 添加至实例end
4) for
${v_j} \in {G_{{\text{redundant}}}}$ do通过式(1)将约束
$ {x_{{v_j}}} = 0 $ 添加至实例end
5) 通过执行步骤3)~4),使用线性整数规划问题ILP计算
$ G = (V,E) $ 的MDS解决方案$M$ 6) for
${v_i} \in M$ doif
${v_i} \notin {G_{{\text{critical}}}}$ then 在步骤5)的函数约束基础上新建一个线性整数规划问题ILP实例
$ {I_{{v_i}}} $ ,加入节点${v_i}$ 的约束${x_{{v_i}}} = 0$ ,得到解决方案$ {M_{{v_i}}} $ if
$ {I_{{v_i}}} $ 没有可行解或$\left| {{M_{{v_i}}}} \right| > \left| M \right|$ (解决方案中节点的个数$\left| {{M_{{v_i}}}} \right|$ 要大于原来解决方案中节点的个数$\left| M \right|$ )then
$ {G_{{\text{critical}}}} \leftarrow {v_i} $ else
$ {G_{{\text{intermittent}}}} \leftarrow {v_i} $ 7) for
${v_j} \in V - M$ doif
${v_j} \notin {G_{{\text{redundant}}}}$ then在步骤5)的函数约束基础上新建一个线性整数规划问题ILP实例
$ {I_{{v_j}}} $ ,加入节点${v_j}$ 的约束${x_{{v_j}}} = 1$ ,得到解决方案$ {M_{{v_j}}} $ if
$ {I_{{v_j}}} $ 没有可行解 or$\left| {{M_{{v_j}}}} \right| > \left| M \right|$ then
$ {G_{{\text{redundant}}}} \leftarrow {v_j} $ else
$ {G_{{\text{intermittent}}}} \leftarrow {v_j} $ 8) return
${G_{{\text{critical}}}}$ ,$ {G_{{\text{redundant}}}} $ ,$ {G_{{\text{intermittent}}}} $ 利用控制分类模型,将蛋白质相互作用网络中的基因从复杂的相互作用关系中分类出3部分,每部分集合中的基因之间对网络控制的作用相同。
-
文献[19]提出的中心性–致命性规则指出,蛋白质相互作用网络中高度连接的蛋白质往往是必不可少的。中心性是衡量网络节点重要性的重要指标,能够刻画节点在网络中的地位。如度中心性描述了一个节点对其他节点的直接影响;介数中心性刻画了经过一个节点的最短路径数,表明这个节点对网络最短传输的控制力;接近中心性反映了一个节点与其他节点之间的接近程度,此节点可以通过邻居节点能够迅速地覆盖到整个网络。综合中心性是通过加权的方法,将度中心性、介数中心性、接近中心性归一化,并结合起来衡量节点在网络中的综合表现。为了精确地筛选肿瘤基因集中的更为关键的基因,本文利用综合中心性来衡量基因对生物网络的重要程度,并对从蛋白质相互作用网络中分离出来的肿瘤基因集进行了排序。相关中心性的定义为:
$$ {C_d}\left( {{v_i}} \right) = \frac{{{k_i}}}{{N - 1}} $$ (3) 式中,
${C_d}\left( {{v_i}} \right)$ 表示节点${v_i}$ 的度中心性;${k_i}$ 表示节点${v_i}$ 的邻居节点的数目;$N$ 表示节点个数;$ N - 1 $ 指节点可能的最大度值。$$ {C_b}\left( {{v_i}} \right) = \sum\limits_{s \ne t \ne i \in V} {\frac{{{\sigma _{s,t}}\left( {{v_i}} \right)}}{{{\sigma _{s,t}}}}} $$ (4) 式中,
${C_b}\left( {{v_i}} \right)$ 表示节点${v_i}$ 的介数中心性;${\sigma _{s,t}}$ 表示节点$s$ 与节点$t$ 之间最短路径总数;${\sigma _{s,t}}\left( i \right)$ 表示节点$s$ 与节点$t$ 之间经过节点${v_i}$ 的最短路径数目。$$ {C_{{\rm{cl}}}}\left( {{v_i}} \right) = \frac{N}{{\displaystyle \sum\limits_{j = 1}^N {d({v_i},{v_j})} }}\;\;{i \ne j} $$ (5) 式中,
${C_{{\rm{cl}}}}\left( {{v_i}} \right)$ 表示节点${v_i}$ 的接近中心性;$d( {{v_i},{v_j}} )$ 表示节点${v_i}$ 与节点${v_j}$ 的距离。$$ {C_{{\text{intgr}}}}\left( {{v_i}} \right) = \frac{1}{3}\left[\frac{{{C_d}\left( {{v_i}} \right)}}{{{C_{d,\max }}}} + \frac{{{C_b}\left( {{v_i}} \right)}}{{{C_{b,\max }}}} + \frac{{{C_{{\rm{cl}}}}\left( {{v_i}} \right)}}{{{C_{{\rm{cl}},\max }}}}\right] $$ (6) 式中,
${C_{{\rm{intgr}}}}\left( {{v_i}} \right)$ 表示节点${v_i}$ 的综合中心性;$ {C_{d,\max }} $ ,$ {C_{b,\max }} $ ,${C_{{\rm{cl}},\max }}$ 分别为$ \{ {C_d}\} $ ,$\{ {C_b}\}$ ,$\{ {C_{{\rm{cl}}}}\}$ 中的最大值。 -
本文首先整合了多个人类蛋白–蛋白互作数据库的PPI数据(包含InnateDB[20]、PINA[21]、BioGrid[22]及HINT[23]等),删除其中重复的边和自环,构成了由15474个蛋白形成的170631条边的网络。本文从TCGA数据库[24]中收集了包括乳腺癌(BRCA)、肾透明细胞癌(KIRC)、肺腺癌(LUAD)、结肠腺癌(COAD)和头颈鳞状细胞癌(HNSC)在内的5种癌症类型及其相应的正常样本的RNA-Seq (RPKM)数据,对于每个基因对,计算其共表达的皮尔森相关系数及相应的p值。将不同的癌症类型的共表达关系嵌入到PPI网络中,选择显著的连边(p<0.05)构成癌症特异性蛋白–蛋白网络[25]。
-
本文从WiKiPathways代谢通路数据库[26]中收集了上述5种癌症类型的生物通路数据集,并利用一些已知与癌症相关的生物通路,进一步得到每个与癌症相关的生物通路的基因集,这些基因对各生物功能起着调控作用。
-
本文收集了SMG (significantly mutated genes) [3], CGC (cancer gene census)[27]这两个肿瘤基因集,这些基因集的基因在癌症变化发展中所起到的重要作用都已被广泛证实。
-
本文从TCGA数据库中下载了上述5种癌症类型的肿瘤病人的体细胞突变数据。
-
本文对TCGA数据库中的5种癌症类型的蛋白质相互作用数据进行了处理,从中提取出了具有显著意义的连边(即p<0.05),并利用最大连通子图的定义,剔除了一些在网络中最大连通子图之外的小簇基因集,重构了蛋白质相互作用网络,各个网络的统征如表1所示。
表 1 肿瘤不同阶段的最大连通子图的节点连边情况
癌症类型 节点数量/个 边数量/个 Normal
阶段Tumor
阶段Normal
阶段Tumor
阶段乳腺癌(BRCA) 13126 13412 96108 107707 结肠癌(COAD) 12047 12849 66246 81720 头颈癌(HNSC) 12110 13263 65594 96629 肾透明细胞癌(KIRC) 12917 13516 78760 106101 肺腺癌(LUAD) 11838 12866 72095 87851 为了从各个阶段复杂的蛋白质相互作用对中,找到对肿瘤发展具有关键影响的基因集,本文利用复杂网络控制理论中的最小控制集模型,并通过控制分类模型将各癌症网络各阶段的基因进行了分类,研究这些基因对网络产生的具体影响。通过方法1.3和1.4,得出了各癌症网络各阶段的3种类型的基因集:关键(critical)基因集、间歇性(intermittent)基因集、冗余(redundant)基因集,其包含的基因情况如表2所示。从最小控制集模型和控制分类模型的定义中,可以了解到关键基因集中的基因是满足所有最小控制集配置方案的集合,即不管最小控制集的配置如何变化,这些基因始终包含在控制集中,其他非控制集中的基因都可以通过这些基因相互作用而到达。因此,关键基因集这一分类更有可能符合对肿瘤关键基因的研究预测集合的预期,于是本文提出了一个猜想:关键基因集中的基因能够控制影响肿瘤的变化发展,在肿瘤突变中较为显著。
表 2 各肿瘤不同阶段的基因分类个数
癌症类型 关键(critical)基因数量 间歇性(intermittent)基因数量 冗余(redundant)基因数量 Normal 阶段 Tumor 阶段 Normal 阶段 Tumor 阶段 Normal 阶段 Tumor 阶段 乳腺癌(BRCA) 714 737 2880 2654 9532 10021 结肠癌(COAD) 732 769 3177 2884 8138 9176 头颈癌(HNSC) 786 760 2994 2855 8330 9648 肾透明细胞癌(KIRC) 743 707 3055 2867 9119 9942 肺腺癌(LUAD) 749 735 2911 2978 8178 9153 -
为了验证本文的猜想,即关键基因集所包含的基因对癌症发展的影响显著明显于其他两种类型的基因集,本文将获取的癌症相关的生物通路数据集和显著基因集与这3种类型的基因集进行了富集分析。显著基因集包含了CGC基因集和SMG基因集,它们所涉及的基因已被广泛地被证实对癌症具有重要影响;生物通路数据集中包含了多个与癌症相关的生物功能作用,每个功能都有与其作用相关的基因组。结合并利用上述数据,分析3种类型的基因集合在其中的富集情况,评估它们预测癌症关键基因的性能。本文从网络中随机生成了10 000次与3种类型基因集同等大小的集合作为参考,并与获取的数据集做了交集,来衡量这3种类型基因集相对于随机基因集的显著程度。本文利用了Z-Score值来表示3种基因集的显著性,图2显示了肿瘤样本Tumor的3种类别的基因集在共有的7种生物通路功能和显著基因集(SMG和CGC)的富集程度(正常样本Normal的富集情况与其类似)。图中横坐标中C表示critical;I表示intermittent;R表示redundant,纵坐标中AG表示angiogenesis;CC表示cell cycle;DDR表示DNA damage response;ICP表示integrated cancer pathway; MR表示mismatch repair; SPIG表示signaling pathways in glioblastoma;TN表示TP53 network。从图中可以看到:关键基因集具有统计学意义,富集度明显优于间歇性基因集、冗余基因集。结果表明,关键基因集能够很好地解释其在生物通路(血管生成、细胞周期、DNA损伤反应、综合癌症通路、胶质母细胞瘤信号通路、TP53通路)上的重要程度,以及富含癌症驱动基因的良好表现。在本阶段工作中,本文从复杂的蛋白质相互作用网络中初步筛选出了肿瘤关键基因集,使得本文可以从该基础上进一步地缩小规模,预测更为重要的肿瘤关键基因。

图 2 5种癌症Tumor阶段各类集合富集分析
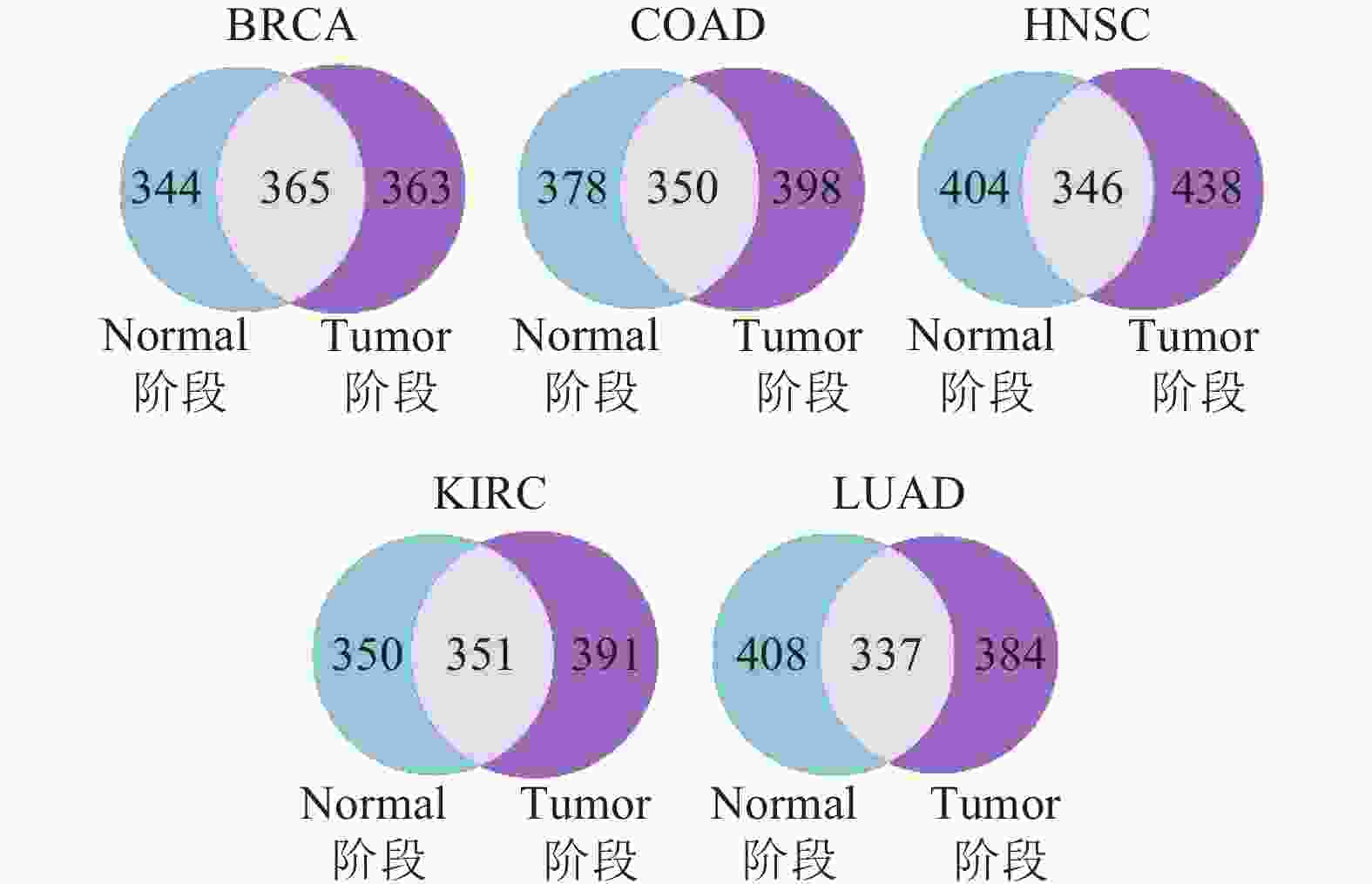
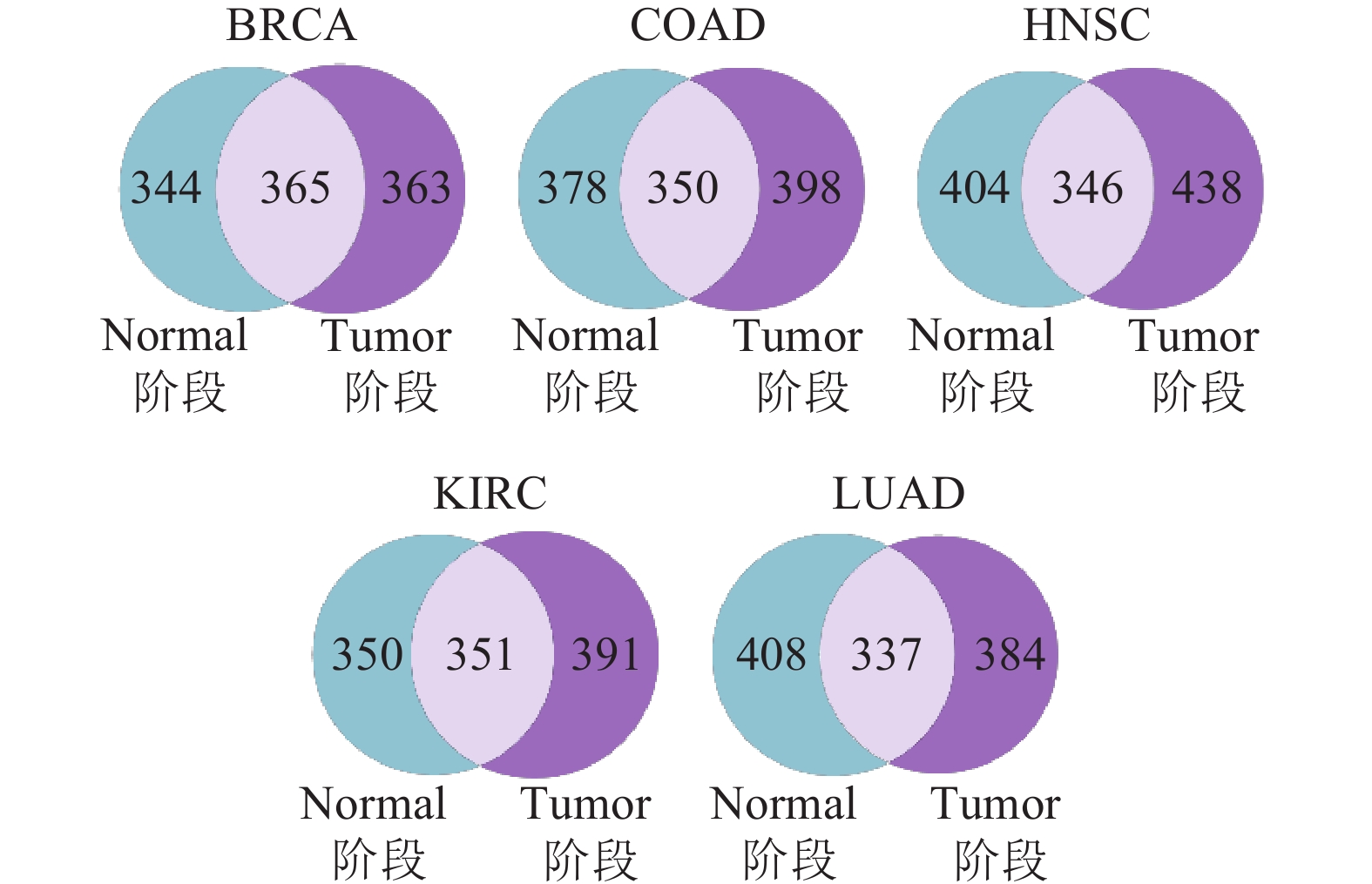
图 3 5种癌症类型Normal阶段和Tumor阶段关键基因集的韦恩图
对各类癌症的多阶段的关键基因集的作用情况进行统计,如图3所示,发现上述分类统计的肿瘤关键基因在整体中占比相近。为了进一步挖掘可预测的关键基因,本文利用显著基因集(SMG, CGC)对这3个部分进行富集占比分析,评估各个部分在肿瘤的突变过程中的作用效果,最终筛选出对肿瘤突变产生至关影响的部分。图4给出了各癌症不同的集合在这两个显著基因集中的不同占比情况,可以发现:仅在Tumor阶段为关键基因的集合(Tumor-Normal)更具有研究意义,其存在于显著基因集的占比在大多数情况下(BRCA、COAD、HNSC、KIRC、LUAD)都要优于另外两个交集集合。相比显著基因集在Normal或者Tumor中的占比,除了个别情况外(存在于癌症的COAD、HNSC集合中显著基因集CGC的占比),仅在Tumor阶段为关键基因的集合所存在的比例仍优于前者。考虑到单阶段(Normal或Tumor)的关键基因集,不能反应肿瘤在突变过程的变化,解释不了其基因的突变以及对整个癌症的影响趋势,综合Normal、Tumor两个阶段选择的关键基因集的研究要优于单阶段的关键基因集。图4的COAD、HNSC中,Normal阶段的关键基因集与仅在Tumor阶段为关键基因的集合的比例相差在0.5%内,但这并不影响本文对结果(仅在Tumor阶段为关键基因的集合)的选择。

图 4 各癌症各阶段各部分的关键基因集与SMG、CGC基因的占比情况
-
上述研究工作对肿瘤关键基因集进行了筛选,最终选择了仅存在于Tumor阶段(Tumor-Normal)的肿瘤关键基因集,用于对驱动癌症发展及突变的关键基因的预测。本文综合了复杂网络的度量指标,利用综合中心性对最终的关键基因集按照分数进行降序排列,排名较高的基因更有可能驱动癌症的发展,可作为潜在的靶点基因。接着利用SMG、 CGC显著基因集,排除了已知被广泛证实有驱动作用的一些关键基因。最后,对剩下的关键基因进行了分析,同时来验证本文所预测的未知的驱动基因在癌症发展变化过程中的显著表现。
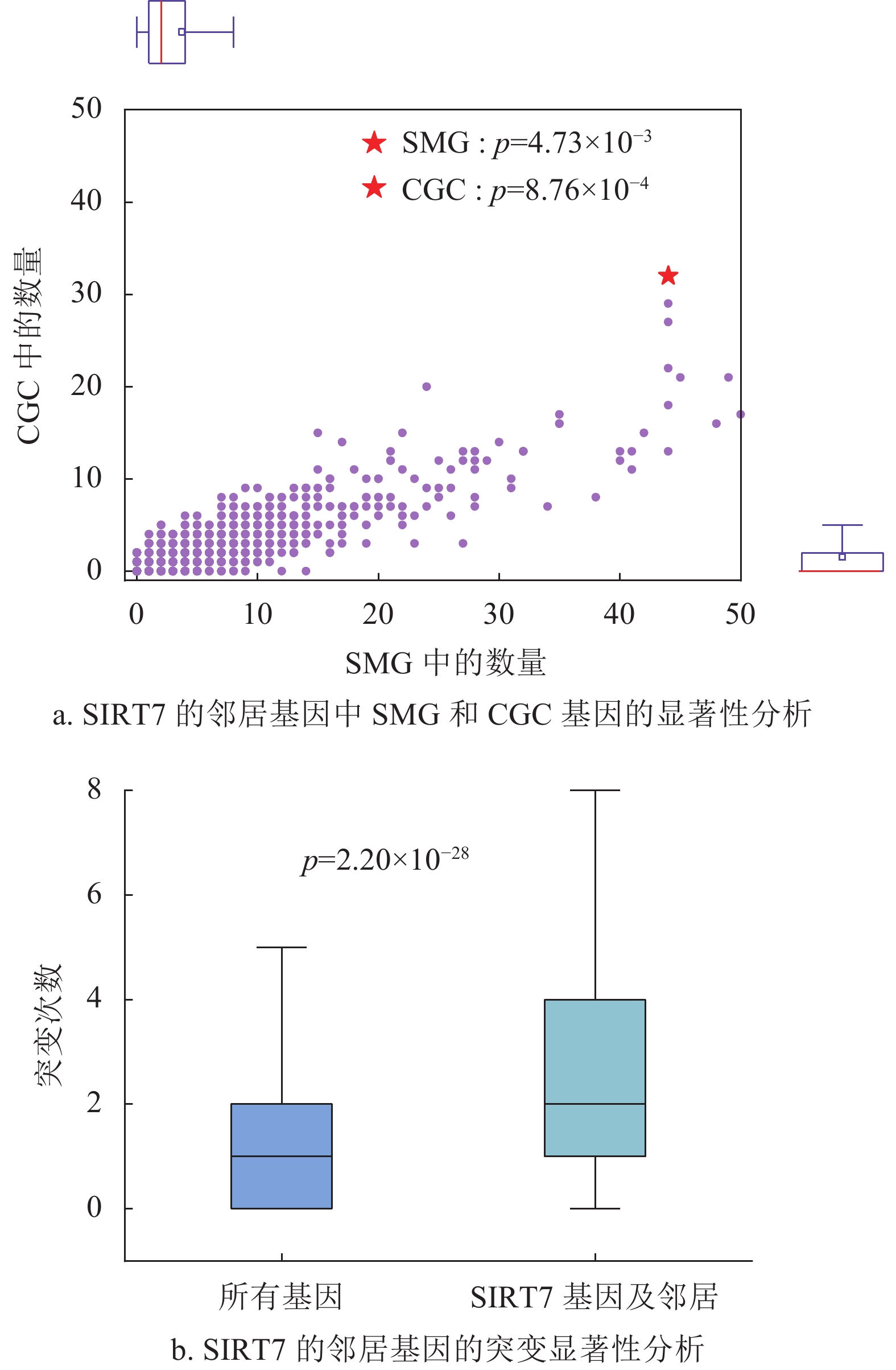
在乳腺癌(BRCA)的蛋白质相互作用网络中,本文选取了综合中心性排名靠前的SIRT7基因作为验证。图5a描述了SIRT7的邻居节点中SMG、CGC基因的富集程度。为了排除一般基因(通常表现为低度)对结果的影响,实验选取了排名在前40%的度较高的基因作为参考标准。其中图5a中的红色标记为SIRT7的邻居中SMG和CGC的基因个数,紫色标记则为其他参考基因集与显著基因集的交集基因分布情况。本文将交集个数大于等于观察值的基因与总体的占比作为显著性检验(p)的衡量标准。从结果可以看出,SIRT7基因的邻居节点相对于其他基因的邻居节点,在两个显著基因集中都显著富集(SMG下p为4.73×10−3,CGC下p为8.76×10−4)。实验证明,SIRT7对肿瘤网络的控制有着重要作用,不仅从它作用性质的角度,而且可以通过它本身的突变带动其周围邻居基因的突变,同时这些邻居基因中富集了与癌症相关的显著基因。图5b展示了SIRT7和它的邻居基因的总体突变分布,其突变分布在T检验中具有统计学意义(p=2.20×10−28),突变频率明显大于整体,在癌症中具有较高的突变频次。先前有实验研究表明SIRT7的表达可作为乳腺癌的预后生物标志物[28-29]。深圳大学健康科学中心的研究人员发现,SIRT7与乳腺癌肺转移相关,SIRT7通过转化因子−β信号调节EMT,能够抑制乳腺癌向肺部的转移,提供了有效的乳腺癌转移治疗策略[28]。深圳大学附属第一医院的研究人员发现,SIRT7可能是乳腺癌瘤体免疫浸润相关的预后生物标志物[29]。
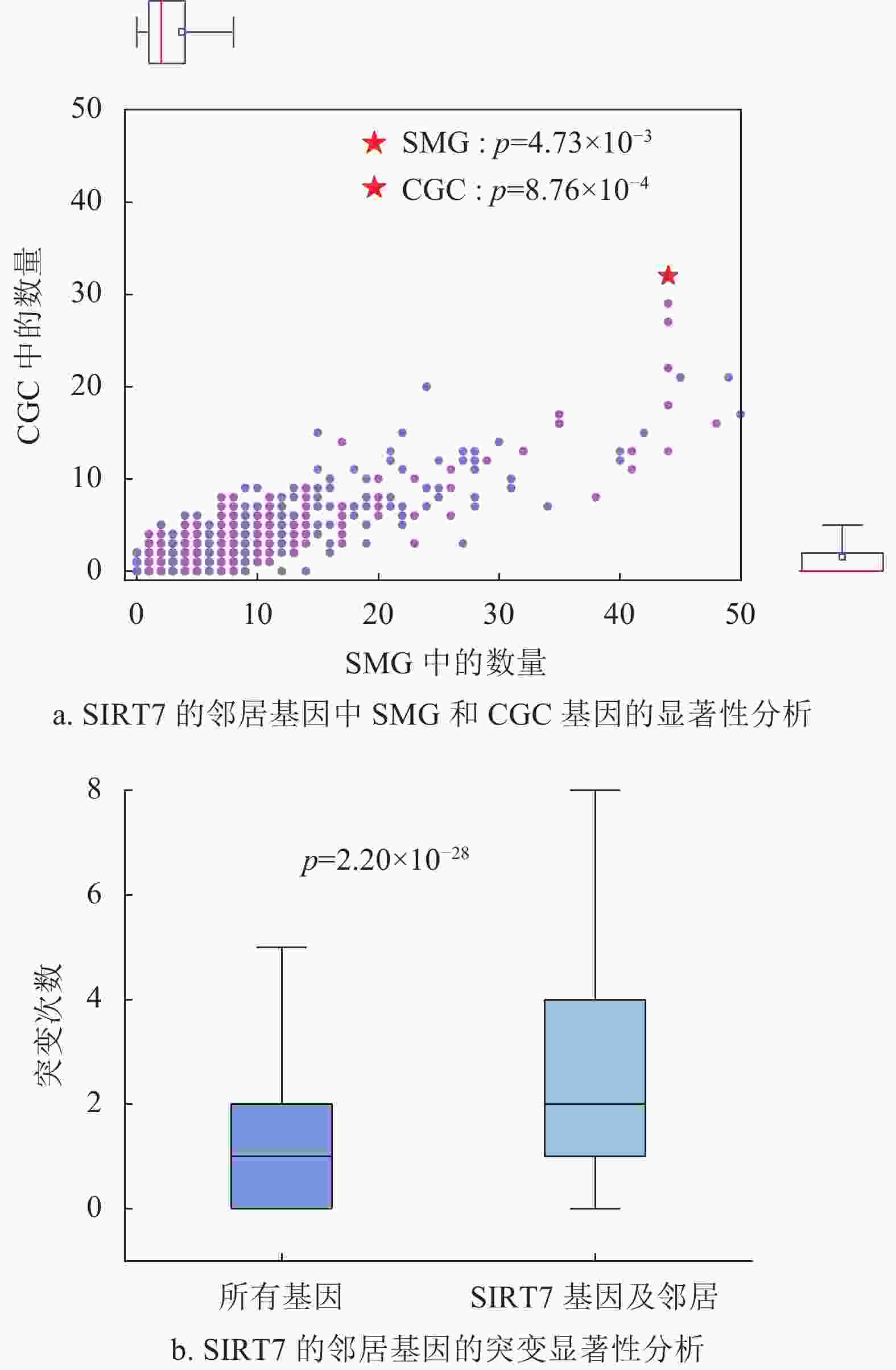
图 5 乳腺癌中的SIRT7基因的重要性分析
在头颈部鳞状细胞癌(HNSC)的蛋白质相互作用网络中,本文选择了ETS1基因作为验证,图6a描述了ETS1基因的邻居为SMG、CGC基因的情况,结果表明ETS1的邻居显著基因数要明显多于所参考的基因集(SMG下p为1.66×10−2, CGC下p为4.56×10−2),这也验证了基因的度中心性较高的基因更有可能与更多的显著基因相邻。ETS1基因在癌症中的表达变化,影响着与其相关的显著基因,在发生癌变的过程中担任着重要角色。图6b显示了ETS1基因和它的邻居相对于蛋白质相互作用网络中所有基因及其邻居的表现情况,结果表明ETS1基因以及它的邻居的突变分布要高于网络整体水平(p=3.49×10−4),可作为致头颈癌产生癌变的关键基因。已有研究表明ETS1可以作为治疗头颈癌的关键靶点[30-31]。美国纽约州立大学的研究人员发现ETS1是头颈部鳞状细胞癌的生物标志物,它在头颈部鳞状细胞癌中的过度表达与预后不良相关,并且是关键上皮性向间质转化(EMT)基因的主要调节因子,为肿瘤亚型特异性的靶向治疗提供新途径[30]。文献[31]发现SRC-ETS1生存通路的上调与头颈部鳞状细胞癌HNSC的细胞增殖、存活、迁移、侵袭和顺铂耐药有关。
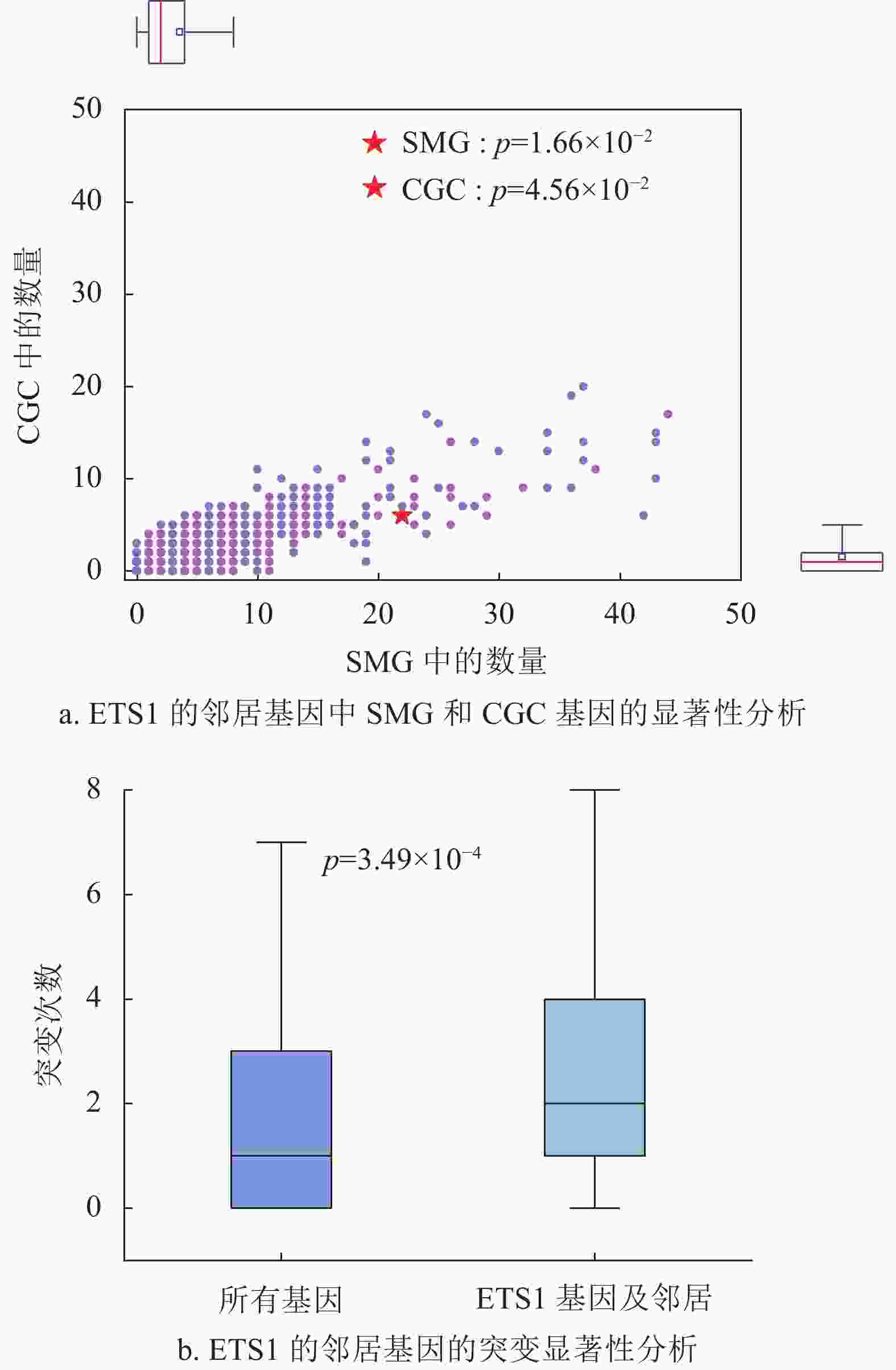
图 6 头颈癌中的ETS1基因的重要性分析
-
网络控制理论能够揭示预测蛋白质互作网络的相互作用机制,能够从复杂的生物网络中识别出肿瘤关键基因,为癌症的药物设计及预后生物标志物预测提供了很好的借鉴。本文利用复杂网络控制理论的方法对来自TCGA数据库中5种癌症网络肿瘤基因进行了分类筛选,并结合肿瘤基因在两个阶段中的蛋白质网络相互作用情况,进一步确认了肿瘤的关键基因集合。利用网络的综合中心性,除去已知被证实为显著基因,对其他肿瘤关键基因集进行排序,得到潜在的肿瘤关键基因。通过验证研究分析,在网络中具有生物学统计意义的关键基因在肿瘤发展过程中的研究可能有着重要影响,它们能够作为药物靶点或者作为预后生物标志物,推动癌症的治疗及防控的研究。
本文利用复杂网络控制理论和基因网络分析,结合生物医疗数据对肿瘤的关键基因进行预测,为网络控制在生物医疗方面的研究提供了良好的思路。但是本研究中只考虑了Normal和Tumor两个阶段的癌症蛋白质相互作用网络,如果进一步地结合Tumor不同时期的数据细化肿瘤关键基因的识别,可以更好地进行肿瘤演化方面的分析,这也是未来工作中的研究方向。
Predicting the Critical Tumor Genes Based on Complex Network Control Theory
-
摘要: 复杂网络控制能够捕获整个网络的状态,使得从海量的蛋白质相互作用数据中找到潜在的肿瘤致病基因成为可能。该文利用复杂网络控制理论探究肿瘤关键基因,对5种癌症相关的蛋白质–蛋白质相互作用网络,通过网络最小控制集方法,选取始终处于最小控制集(minimum dominating set, MDS)的基因作为候选关键基因。利用肿瘤相关的生物通路数据和已被证实的肿瘤基因数据,采用富集分析证明了该方法的有效性。构建网络综合中心性指标,对候选关键基因进行排序。进而针对不同的癌症类型,挑选排在前面的候选基因(非已知重要基因集的基因)作为最终的预测基因,基于网络结构和体细胞突变数据分析,对其作为生物标志物的有效性进行验证。该研究在一定程度上为复杂网络控制理论在生物医学中的应用提供了思路。Abstract: Network control theory can capture the state of the whole network, which makes it possible to find potential tumor-causing genes from massive complicated protein-protein interaction data. To explore the key genes of tumors, complex network control theory is applied in this paper to analyze the protein-protein interaction networks with five different kinds of cancers. We mine the minimum dominating set (MDS) of the network and select the genes that always belong to the MDS as the candidate key genes. Using the tumor related pathways and essential tumor gene sets, we find that the candidate key genes are clustered in these gene sets, which indicate the effectiveness of the methods based on MDS. In addition, a comprehensive centrality method is proposed to rank the candidate genes with this method, and then the top ranked genes are selected as the candidate biomarkers. Furthermore, we evaluate the probability of top ranked genes being biomarkers according to the network structural analysis and the enrichment of the somatic mutation. In summary, this study may shed light on the application of complex network control theory in biomedicine.
-
Key words:
- biomarkers /
- complex network control /
- key genes /
- protein interaction /
- tumor
-
表 1 肿瘤不同阶段的最大连通子图的节点连边情况
癌症类型 节点数量/个 边数量/个 Normal
阶段Tumor
阶段Normal
阶段Tumor
阶段乳腺癌(BRCA) 13126 13412 96108 107707 结肠癌(COAD) 12047 12849 66246 81720 头颈癌(HNSC) 12110 13263 65594 96629 肾透明细胞癌(KIRC) 12917 13516 78760 106101 肺腺癌(LUAD) 11838 12866 72095 87851  下载: 导出CSV
下载: 导出CSV
表 2 各肿瘤不同阶段的基因分类个数
癌症类型 关键(critical)基因数量 间歇性(intermittent)基因数量 冗余(redundant)基因数量 Normal 阶段 Tumor 阶段 Normal 阶段 Tumor 阶段 Normal 阶段 Tumor 阶段 乳腺癌(BRCA) 714 737 2880 2654 9532 10021 结肠癌(COAD) 732 769 3177 2884 8138 9176 头颈癌(HNSC) 786 760 2994 2855 8330 9648 肾透明细胞癌(KIRC) 743 707 3055 2867 9119 9942 肺腺癌(LUAD) 749 735 2911 2978 8178 9153
下载: 导出CSV
-
[1] 刘宗超, 李哲轩, 张阳, 等. 2020全球癌症统计报告解读[J]. 肿瘤综合治疗电子杂志, 2021, 7(2): 1-14. LIU Z C, LI Z X, ZHANG Y, et al. Interpretation on the report of global cancer statistics 2020[J]. Journal of Multidisciplinary Cancer Management, 2021, 7(2): 1-14. [2] BAILEY M H, TOKHEIM C, PORTA-PARDO E, et al. Comprehensive characterization of cancer driver genes and mutations[J]. Cell, 2018, 174(4): 1034-1035. doi: 10.1016/j.cell.2018.07.034 [3] CHENG F X, LU W Q, LIU C, et al. A genome-wide positioning systems network algorithm for in silico drug repurposing[J]. Nature Communications, 2019, 10(1): 1-14. doi: 10.1038/s41467-018-07882-8 [4] NUSSINOV R, JANG H, TSAI C J, et al. Precision medicine review: Rare driver mutations and their biophysical classification[J]. Biophysical Reviews, 2019, 11(1): 5-19. doi: 10.1007/s12551-018-0496-2 [5] WEINSTEIN J N, COLLISSON E A, MILLS G B, et al. The cancer genome atlas pan-cancer analysis project[J]. Nature Genetics, 2013, 45(10): 1113-1120. doi: 10.1038/ng.2764 [6] 涂超峰, 綦鹏, 李夏雨, 等. 肿瘤异质性: 精准医学需破解的难题[J]. 生物化学与生物物理进展, 2015, 42(10): 881-890. TU C F, QI P, LI X Y, et al. Tumor heterogeneity: The challenge of prediction medicine[J]. Progress in Biochemistry and Biophysics, 2015, 42(10): 881-890. [7] MERTINS P, MANI D R, RUGGLES K V, et al. Proteogenomics connects somatic mutations to signalling in breast cancer[J]. Nature, 2016, 534(7605): 55-62. doi: 10.1038/nature18003 [8] SHI Y, GAO W, LYTLE N K, et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring[J]. Nature, 2019, 569(7754): 131-135. doi: 10.1038/s41586-019-1130-6 [9] HU J X, THOMAS C E, BRUNAK S. Network biology concepts in complex disease comorbidities[J]. Nature Reviews Genetics, 2016, 17(10): 615-629. doi: 10.1038/nrg.2016.87 [10] LIU C, MA Y F, ZHAO J, et al. Computational network biology: Data, models, and applications[J]. Physics Reports, 2020, 846: 1-66. doi: 10.1016/j.physrep.2019.12.004 [11] 侯绿林, 老松杨, 肖延东, 等. 复杂网络可控性研究现状综述[J]. 物理学报, 2015, 64(18): 481-491. HOU L L, LAO S Y, XIAO Y D, et al. Recent progress in controllability of complex network[J]. Acta Physica Sinica, 2015, 64(18): 481-491. [12] LIU Y Y, SLOTINE J J, BARABASI A L. Controllability of complex networks[J]. Nature, 2011, 473(7346): 167-173. doi: 10.1038/nature10011 [13] NACHER J C, AKUTSU T. Dominating scale-free networks with variable scaling exponent: Heterogeneous networks are not difficult to control[J]. New Journal of Physics, 2012, 14(7): 073005. doi: 10.1088/1367-2630/14/7/073005 [14] VINAYAGAM A, GIBSON T E, LEE H J, et al. Controllability analysis of the directed human protein interaction network identifies disease genes and drug targets[J]. Proceedings of the National Academy of Sciences, 2016, 113(18): 4976-4981. doi: 10.1073/pnas.1603992113 [15] WUCHTY S. Controllability in protein interaction networks[J]. Proceedings of the National Academy of Sciences, 2014, 111(19): 7156-7160. doi: 10.1073/pnas.1311231111 [16] ZHANG X F, OU-YANG L, ZHU Y, et al. Determining minimum set of driver nodes in protein-protein interaction networks[J]. BMC Bioinformatics, 2015, 16: 146. doi: 10.1186/s12859-015-0591-3 [17] NACHER J C, AKUTSU T. Analysis of critical and redundant nodes in controlling directed and undirected complex networks using dominating sets[J]. Journal of Complex Networks, 2014, 2(4): 394-412. doi: 10.1093/comnet/cnu029 [18] ISHITSUKA M, AKUTSU T, NACHER J C. Critical controllability in proteome-wide protein interaction network integrating transcriptome[J]. Scientific Reports, 2016, 6: 23541. doi: 10.1038/srep23541 [19] JEONG H, MASON S P, BARABASI A L, et al. Lethality and centrality in protein networks[J]. Nature, 2001, 411(6833): 41-42. doi: 10.1038/35075138 [20] BREUER K, FOROUSHANI A K, LAIRD M R, et al. InnateDB: Systems biology of innate immunity and beyond—recent updates and continuing curation[J]. Nucleic Acids Research, 2013, 41(D1): D1228-D1233. doi: 10.1093/nar/gks1147 [21] COWLEY M J, PINESE M, KASSAHN K S, et al. PINA v2.0: Mining interactome modules[J]. Nucleic Acids Research, 2012, 40(D1): D862-D865. doi: 10.1093/nar/gkr967 [22] STARK C, BREITKREUTZ B J, REGULY T, et al. BioGRID: A general repository for interaction datasets[J]. Nucleic Acids Research, 2006, 34(suppl 1): D535-D539. [23] DAS J, YU H. HINT: High-quality protein interactomes and their applications in understanding human disease[J]. BMC Systems Biology, 2012, 6(1): 1-12. doi: 10.1186/1752-0509-6-1 [24] The National Institutes of Health. The cancer genome atlas[EB/OL]. [2019-09-20]. https://portal.gdc.cancer.gov/. [25] YAO X. MDS classification[EB/OL]. [2021-07-10]. https://github.com/Spainstaking/MDS_Classification. [26] KELDER T, PICO A R, HANSPERS K, et al. Mining biological pathways using WikiPathways web services[J]. PloS One, 2009, 4(7): e6447. doi: 10.1371/journal.pone.0006447 [27] TOKHEIM C J, PAPADOPOULOS N, KINZLER K W, et al. Evaluating the evaluation of cancer driver genes[J]. Proceedings of the National Academy of Sciences, 2016, 113(50): 14330-14335. doi: 10.1073/pnas.1616440113 [28] TANG X, SHI L, XIE N, et al. SIRT7 antagonizes TGF-beta signaling and inhibits breast cancer metastasis[J]. Nature Communications, 2017, 8(1): 318. doi: 10.1038/s41467-017-00396-9 [29] HUO Q, LI Z, CHENG L, et al. SIRT7 is a prognostic biomarker associated with immune infiltration in luminal breast cancer[J]. Frontiers in Oncology, 2020, 10: 621. doi: 10.3389/fonc.2020.00621 [30] GLUCK C, GLATHAR A, TSOMPANA M, et al. Molecular dissection of the oncogenic role of ETS1 in the mesenchymal subtypes of head and neck squamous cell carcinoma[J]. PLoS Genetics, 2019, 15(7): e1008250. doi: 10.1371/journal.pgen.1008250 [31] YANG Z, LIAO J, CARTER-COOPER B A, et al. Regulation of cisplatin-resistant head and neck squamous cell carcinoma by the SRC/ETS-1 signaling pathway[J]. BMC Cancer, 2019, 19(1): 485. doi: 10.1186/s12885-019-5664-7 -
 点击查看大图
点击查看大图
图(6) / 表(2)
计量
- 文章访问数: 4193
- HTML全文浏览量: 1229
- PDF下载量: 48
- 被引次数: 0














































































































