 ISSN
ISSN 


-
抗体(antibody)是指机体内一类能识别外来潜在有害分子的表面并与之特异性结合,从而引发适应性免疫反应的免疫球蛋白(immunoglobulin, Ig)[1]。抗体在脊椎动物的免疫防御系统中起着不可或缺的作用[2],在重大疾病的预防、诊断与治疗中也起着至关重要的作用[3]。在临床实践中,单克隆抗体(monoclonal antibody, mAb)的出现对癌症和自身免疫性疾病的治疗产生了革命性的影响,不少化学小分子不能作用的蛋白成为抗体药物的高效靶标。由于二代测序、噬菌体展示等技术的发展,抗体药物产业进入飞速发展的时代。单克隆抗体的结合特异性、安全性、构象稳定、可制造性等多种特性,使其成为目前最大的一类生物治疗药物[1]。据统计,美国FDA批准上市的抗体药已多达100种[4],全球正在进行I、II期临床试验的抗体药物超过550种,另有79种已进入开发最后阶段[5]。全球抗体药物占据着巨大的市场份额,2018年全球最畅销的药品TOP10中,就有8种是抗体药物,当年抗体的全球市场价值为1152亿美元,预计2025年能达到3000亿美元[6]。无论从临床价值还是产业角度,抗体药物相关的研究在生物医药行业的热度会持续增长。虽然抗体产业发展如此迅猛,但进入临床试验阶段的人源或人源化治疗性抗体最终能够成功开发上市的只有15%左右[7]。不少抗体由于低表达、低稳定性、高聚集、存在交叉或自身相互作用等问题导致研发失败。许多相互依存的因素影响着治疗性抗体的成功开发,而选择具有良好理化特性的候选药能为后续研发奠定良好基础。可开发性(developability)评估[8]的主要目标是严格评估单克隆抗体候选物的生化和生物物理特性,并选择开发风险低的抗体分子。
目前,对抗体生物物理与生物化学特性进行测试已有多种相关实验方法,如反映抗体的粘度、聚集倾向、溶解度的直立单层色谱(SMAC)、体积排除色谱(SEC)、亲和捕获自相互作用纳米颗粒光谱(AC-SINS)、克隆自相互作用生物膜干涉(CSI-BLI)、疏水相互作用色谱(HIC);反应抗体结合特异性的常见抗原或杆状病毒颗粒(BVP)的酶联免疫吸附试验(ELISA)、多特异性试剂结合试验(PSR);反映抗体热稳定性的差示扫描荧光(TmDSF)等[9]。文献[10]对被批准上市或处于II、III期临床试验的137个抗体进行了12种实验测定,给每项实验中表现最差的10%的抗体标记一项缺陷。结果显示,约2/3的上市抗体没有缺陷,而二期临床试验中的抗体约2/3有一项或以上缺陷。这一观察结果提示了大规模测试综合评估抗体理化特性,可作为候选抗体的可开发性评估标志[10]。然而,通过实验评估抗体药物可开发性相关理化性质费事费钱费力。相较而言,计算方法更省时省钱[11-12]。
对抗体理化性质进行预测在近年来迅速成为免疫信息学研究的热点。文献[13]对12种抗体的长期稳定性进行了为期2年的测量,他们计算抗体净电荷、空间聚集属性(spatial aggregation propensity, SAP)分数,从中得出了可开发性指数(developability index, DI),并证明了其与抗体的聚集属性密切相关。然而,DI基于抗体晶体结构或同源建模形成的理论结构进行计算,速度较慢,结果受理论预测模型精度的影响较大。文献[14]仅基于序列预测抗体聚集倾向,最佳模型的AUC为0.76,性能欠佳,且收费使用。文献[15]仅根据抗体序列,采用支持向量机训练了能能预测抗体交叉或自身相互作用的模型CISI,准确率可达到88.20%,该模型可快速高通量地评估影响抗体可开发性的交叉或自身相互作用,缺点是模型中所含的特征数过多,容易导致过拟合,泛化性较弱。
针对上述问题,本文提出一个新的模型,用于预测抗体的交叉或自身相互作用,并提供了免费在线服务工具。该模型可作为抗体可开发性评估流程中的一个环节,以便研究者快速筛选出可开发性较高的候选抗体,加快研究进程,降低研发成本。
-
本文所使用的数据下载自文献[10],包括48条已被批准的和89条处于临床II、III试验的抗体序列信息以及已被广泛运用于抗体理化性质测定的12种实验检测的结果。本文采用多特异性试剂结合试验 (poly-specificity reagent, PSR)、交叉作用色谱 (cross-interaction chromatography, CIC)、克隆自相互作用生物膜干涉测定 (clone self-interaction by biolayer interferometry, CSI-BLI)、亲和捕获自相互作用纳米颗粒光谱 (affinity-capture self-interaction nanoparticle spectroscopy, AC-SINS)4个指标对抗体的交叉或自身相互作用进行综合评估。
-
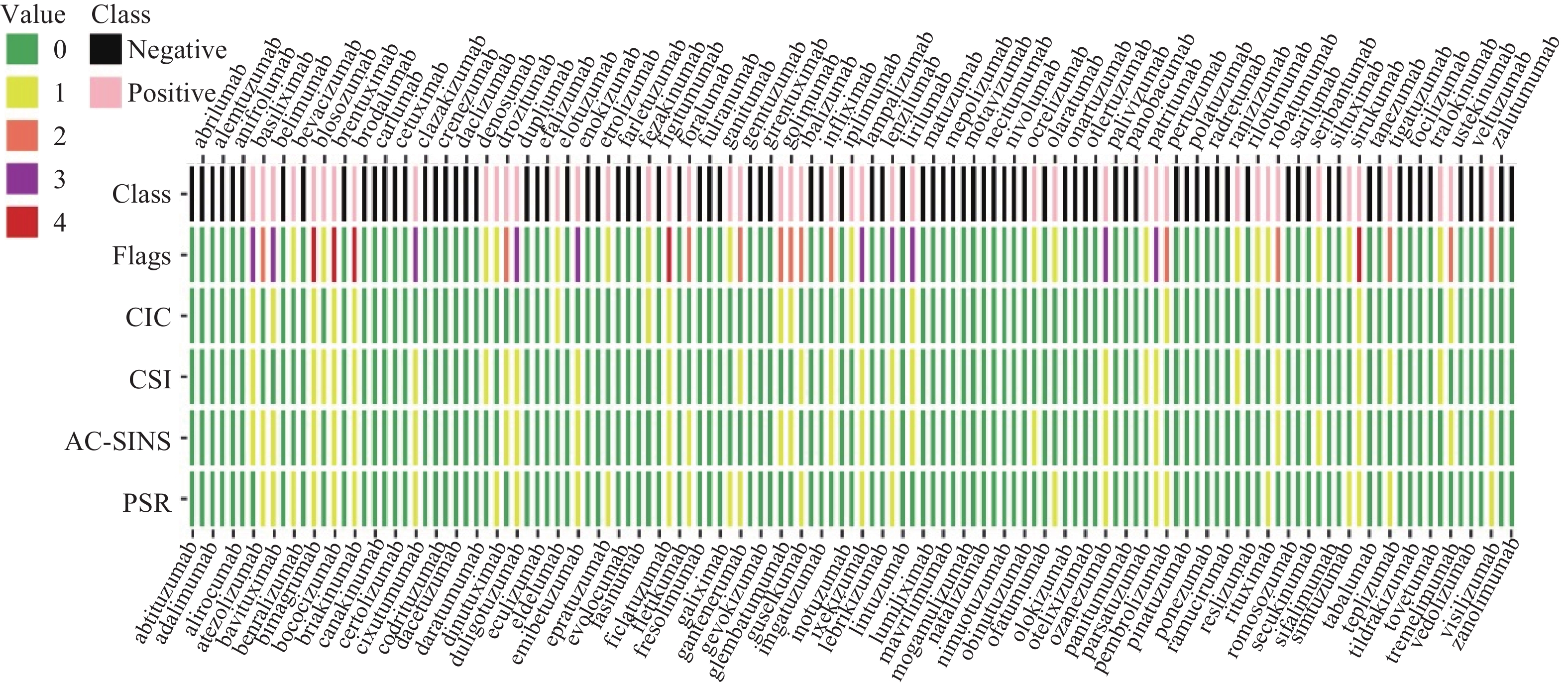
为保证数据的准确性,有冲突记录的6条抗体序列被排除。将抗体重链可变区与轻链可变区连接(重链在前,轻链在后)作为抗体的序列。文献[10]对给每项实验中表现最差的10%的抗体标记一项缺陷,当实验数据超过或低于该阈值时,该抗体则标记为一项缺陷。根据缺陷数,将抗体分为正负样本,至少有一项缺陷的为正样本。4种实验的阈值如表1所示,最终得到85条阴性样本(未显示缺陷标记),46条阳性样本,每条抗体的标记情况详见图1。CIC、CIS、AC-SINS、PSR:1表示该实验方法测出的结果显示抗体有缺陷,0表示没有缺陷。flags:每种缺陷标记之和,大于等于1时抗体为阳性样本,等于0时抗体为阴性样本。在机器学习中,正负样本不平衡是一个常见的问题,多数量样本所含有的信息量较大,会对分类器学习过程造成影响。为解决该问题,85条阴性样本被随机分为包含42、43条抗体的两组,再分别与46条阳性样本组成两个相对平衡的子数据集(group1、group2),分别构建模型,最后集成模型预测抗体的交叉或自身相互作用。
表 1 4种评估抗体交叉或自身相互作用的实验阈值
实验 阈值 单位(标记) PSR (0–1) 0.27 None (>) AC-SINS 11.8 Nanometers (wavelength change) (>) CIC 10.1 Retention time (min) (>) CSI-BLI 0.01 BLI response units (>) 
图 1 每个抗体交叉或自身相互作用的缺陷标记热图
-
目前机器学习技术已越来越多的用于DNA、RNA、蛋白质序列分析,但其输入必须是数值而不能是字符。构建一个高效的预测工具,除了基准数据集可靠之外,使用适当的方法,将序列数据无失真地转为数值表达,以描述他们与结构和功能属性的内在关联也是至关重要的[16]。基于大量实验,本研究选择二肽与期望均值的偏差(dipeptide deviation from expected mean, DDE)[17]来提取序列特征。氨基酸频率与各自期望平均值的偏差值由文献[18]提出,用于确定蛋白质的亲缘关系。编码20种氨基酸的密码子数有所不同,理论上,编码相邻的两个氨基酸的密码子数目越多,该二肽组分出现的频率越大,DDE反应了二肽组分出现的频率与期望值偏差,DDE绝对值越大,说明该二肽组分与理论值偏差较大,能够揭示特定蛋白质序列的潜在标志。DDE通过计算二肽组分
$ {D}_{c} $ 、理论均值$ {T}_{m} $ 和理论方差$ {T}_{v} $ 3个参数构建DDE特征向量,具体计算过程如下:$$ {D}_{C}\left(r,s\right)=\frac{{N}_{rs}}{N-1} \;\; \quad r,s\in A,C,\cdots ,Y $$ (1) 式中,
$ {N}_{rs} $ 是由r和s型氨基酸所代表的二肽的数量;N是蛋白质或肽的长度。$$ {T}_{m}(r,s)=\frac{{C}_{r}}{{C}_{N}}\frac{{C}_{s}}{{C}_{N}} $$ (2) 式中,对于给定的二肽r、s,
$ {C}_{r} $ 是编码第一个氨基酸r的密码子数量;$ {C}_{s} $ 是编码第二个氨基酸s的密码子数量;$ {C}_{N} $ 是密码子总数,除去3个终止密码子,编码氨基酸的密码子数$ {C}_{N} $ =61。根据理论均值$ {T}_{m} $ ,即可算出理论方差$ {T}_{v} $ :$$ {T}_{v}\left(r,s\right)=\frac{{T}_{m}(r,s)(1-{T}_{m}(r,s\left)\right)}{N-1} $$ (3) DDE可被定义为:
$$ \mathrm{D}\mathrm{D}\mathrm{E}\left({r},{s}\right)=\frac{{D}_{C}\left(r,s\right)-{T}_{m}\left(r,s\right)}{\sqrt{{T}_{v}\left(r,s\right)}} $$ (4) 对于400种二肽组成,均计算他们之间DDE,最终可得到一个400维的特征向量,可表示为:
$$ {{\boldsymbol{DDE}}}_{p}=\left\{{\mathrm{D}\mathrm{D}\mathrm{E}}_{i},{\mathrm{D}\mathrm{D}\mathrm{E}}_{i+1},\cdots ,\cdots {\mathrm{D}\mathrm{D}\mathrm{E}}_{n}\right\}\quad{i}=\mathrm{1,2},\cdots ,400 $$ (5) 除此之外,为比较不同的特征提取算法,使用iFeature软件包[19]计算了包括AAC, DPC, TPC, CKSAAP, DDE, GAAC, CKSAAGP, GDPC, GTPC, Moran, Geary, NMBroto, CTDC, CTDT, CTDD, CTriad, KSCTriad, SOCNumber, QSOrder, PAAC 这20种特征描述符,除了上述详细介绍的DDE之外,其余算法的具体过程以及用法的参数解释均在iFeature的操作手册中有详细介绍,这里便不再赘述。
-
特征选择(feature selection, FS)也称特征子集选择,指从已有的所有M个特征中,选择出N(N<M)个特征使得目标最优化。特征数过大的特征集中通常会混杂一些冗余或者不相关特征,会出现维度灾难的问题,加大模型训练复杂度。除此之外,在实际应用中,通常用于研究的样本量较少而特征数较多,容易导致模型过拟合的问题。所以,为了减少特征数量,减低学习难度与过拟合风险,提高模型效率与泛化性,在模型构建前,需要对特征进行选择优化。
目前,研究者已提出许多有效的特征选择方法用于降维。集成学习方法已应用到了包括生物信息在内的众多领域中,该方法结合多个单一技术来解决相同的问题,旨在克服单一技术的弱点,同时保留各个技术的优势[20]。文献[21]基于PageRank算法,开发了一种集成排序的方法MRMD2.0,用于特征排序与降维。MRMD2.0首先计算得出不同的特征排序;然后将所有的排序用有向图表示(a→b表示特征b比特征a重要,得到一个链接列表),使用PageRank得到每个特征的新排名;最后采用序列前向选择,从中筛选出最优特征子集。
-
支持向量机(support vector machine, SVM)是由文献[22]在1995年首次提出的用于分类与回归分析中一种监督式学习模型与相关学习算法。SVM的基本思想是在空间中找出间隔最大的、能正确划分正负样本的超平面。在线性可分的情况下,样本点中与超平面距离最近的点称为“支持向量”,其余的样本点称为“非支持向量”。非支持向量的移动与删除不会对最优超平面产生影响,即支持向量对模型起着决定性的作用。支持向量机能有效解决实际问题中样本较少但特征维度较高的问题,它具有的另一个优势是,在线性不可分的情况下,使用核函数,即使用核变换将原数据隐式映射到新的空间,然后在新空间里用线性方法,找超平面将样本分开。支持向量机灵活多变,深受各个领域的研究者喜爱,目前已应用于(超)文本分类、图像分类、金融预测、生物信息学[23-25]等相关领域。鉴于SVM处理分类问题的出色表现,许多团队基于SVM开发了软件包,以便研究者使用。LIBSVM是文献[26]开发的一个操作简单、快速有效的开源软件包,本课题使用该软件包来构建模型。
-
为了直观地衡量一个分类预测器的好坏,需要引入一些指标参数对模型进行评估,对于分类问题,常用的评价指标有:敏性感(sensitivity, Sn)、特异性(specificity, Sp)、准确率(accuracy, ACC)、马氏相关系数(MCC)等。其中Sn与Sp只能表示对一类样本的预测能力。ACC代表了模型的整体预测准确度。MCC是一个相对平衡的预测评价指标,它综合考虑了TP、TN、FP和FN,避免了样本不平衡偏差。相关的计算公式如下:
$$ \mathrm{S}\mathrm{n}=\frac{{\rm{TP}}}{{\rm{TP}}+{\rm{FN}}} $$ (6) $$ \mathrm{S}\mathrm{p}=\frac{{\rm{TN}}}{{\rm{TN}}+{\rm{FP}}} $$ (7) $$ \mathrm{A}\mathrm{C}\mathrm{C}=\frac{{\rm{TN}}+{\rm{TP}}}{{\rm{TP}}+{\rm{FN}}+{\rm{TN}}+{\rm{FP}}} $$ (8) $$ \mathrm{M}\mathrm{C}\mathrm{C}=\frac{{\rm{TN}}\times {\rm{TP}}-{\rm{FP}}\times {\rm{FN}}}{\sqrt{({\rm{TP}}+{\rm{FP}})({\rm{TP}}+{\rm{FN}})({\rm{TN}}+{\rm{FP}})({\rm{TN}}+{\rm{FN}})}} $$ (9) 此外,本文使用了ROC曲线下面积(area under curve, AUC)作为模型评估的另一项指标。ROC曲线即受试者工作特征曲线(receiver operating characteristic curve),是一个以在不同条件下的1-Sp为横坐标,Sn为纵坐标绘制的二维曲线。AUC的值介于0~1。ROC曲线越靠近左上角,即曲线下面积越大时,模型的预测性能越好。实际情况中,AUC的取值在0.5~1;该值与Sn、Sp、ACC等指标不同,不受预测阈值的影响,是一个更具可比性的反映模型好坏的指标。
-
将用于预测抗体交叉或自身相互作用的数据分为两组(group1、group2),分别进行DDE特征提取,并使用MRMD2.0降维,最终第一组得到的特征数为86、第二组特征数为152。将最优特征子集组成的特征空间作为输入,得到group1与group2两组数据的最优模型参数c、g值(表2),当c与g取最优值时,两个模型的留一交叉检验ACC分别为72.72%与82.02%。接下来分别训练SVM子模型(CISI1与CISI2),表3中列出了两个SVM子模型的详细预测结果。
表 2 两组数据的最优c、g值与模型的准确率
子模型 c g ACC/% CISI1 32.0 0.0078125 72.73 CISI2 512.0 0.0001221 82.02 表 3 两组数据集基于留一法交叉检验的预测模型评估指标
子模型 Sn/% Sp/% ACC/% MCC AUC CISI1 76.09 69.05 72.73 0.4527 0.7257 CISI2 84.78 79.07 82.02 0.6401 0.8193 上述结果中,子模型的准确率最高仅能达到82.02%,本文采用集成方法得到集成模型,提高预测效能,集成策略为:将所有样本作为每个子模型的输入,每个样本得到两个预测结果以及其属于该类别的概率,对两个概率值求平均得到最终的概率均值,作为判断样本类别的标准(阈值为0.5)。如表4所示,当集成模型时,对抗体交叉或自身相互作用的预测准确率能达到96.18%,敏感性能够提升到100.00%,AUC为0.9699,结果表明集成模型是预测抗体自相互作用与交叉相互作用的更优模型。
表 4 抗体交叉或自身相互作用集成模型的预测结果
子模型 Sn/% Sp/% ACC/% MCC AUC CISI2.0 100.00 94.12 96.18 0.9214 0.9699 -
为了确定与抗体交叉或自身相互作用密切相关的特征,表5列出了两个子模型中共同包含的DDE特征,共45个。这些DDE特征中所包含的氨基酸,多为疏水的非极性氨基酸。其中共有11个在正样本组与负样本组之间有着较高的DDE差异比率(>2或<0.5) 如表6所示,结果显示,在正负样本间DDE差异比率最大的二肽组分为TN、FN、GA、NP、WT、DR。在具有交叉或自身相互作用的抗体中,TN的DDE值为负样本组的25.914倍,而NP的DDE值明显降低,仅为负样本组的0.067倍。这说明当表6中列出的二肽组分的出现的频率偏离了期望值时,抗体更易出现交叉或自身相互作用缺陷,在开发前期需要尽早淘汰。
表 5 CISI1与CISI2共有的DDE特征
ER PY MH RW EN DR GA LW CA IN DD VA YD RA GM FS CG GG VT YI YW DK LA WT GS FM TN LH TL WF KA FN NT SI NP IY TV HW IT YF EI TM NS KK HQ 表 6 正样本组与负样本组的平均DDE差异比率
二肽组分 DDE比率(绝对值) TN 25.91415 FN 6.483849 GA 6.234574 IN 3.371006 MH 2.639173 YI 2.381572 TM 0.460651 IT 0.205809 DR 0.168615 WT 0.112033 NP 0.067459 -
为了更为全面地评价不同特征提取算法的预测效果,使用了iFeature包中的其余19种方法对相同分组的数据进行特征提取,MRMD2.0筛选特征子集,建立SVM模型,同样采用投票策略集成预测器,从特征维度,分类器效能方面比较不同的特征提取算法对识别抗体交叉或自身相互作用的影响。
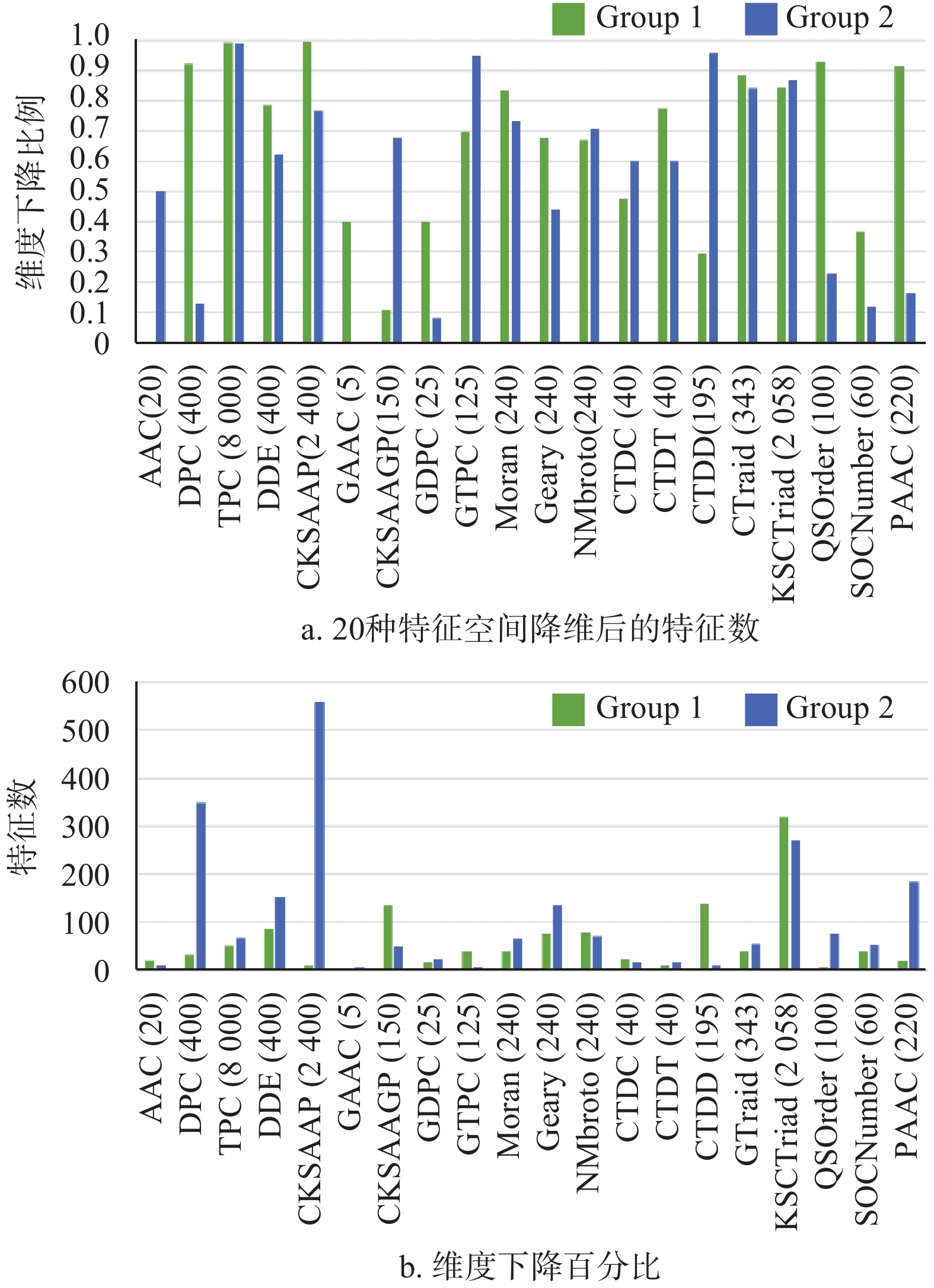
MRMD2.0对每类特征进行降维后的结果如图2所示,图中横坐标括号里为原始特征维度。仅从特征数量看,在预测精度提升的前提下,多数的特征在两个子数据集中都被减少了60%以上,如:TPC、DDE、CKSAAP、CTriad、GTPC、KSCTriad。部分特征,如CKSAAP、CKSAAGP,存在着在两个数据集中维度相差较大的情况,同样说明这些特征容易受样本的影响,缺乏泛化性。
好的特征不仅在数量上要少,同时还能保证模型的效能。20种特征提取算法得到的数据建立集成模型的具体预测结果如表7。基于DDE特征得到模型的Sn、Sp、ACC、MCC与AUC均为最高,因此,在综合特征维度以及集成分类器的分能效能考虑之后,认为DDE这类特征能够作为预测抗体的交叉或自身相互作用的重要标志。

图 2 MRMD2.0降维结果
表 7 20种特征提取算法构建的集成预测器的结果
特征 Sn/% Sp/% ACC/% MCC AUC DDE 100.00 94.12 96.18 0.9214 0.9699 DPC 100.00 82.35 88.55 0.7880 0.9157 KSCTriad 100.00 83.53 89.31 0.8002 0.9157 CKSAAGP 100.00 80.00 87.02 0.7643 0.8976 GDPC 100.00 80.00 87.02 0.7643 0.8976 CTriad 100.00 78.82 86.26 0.7527 0.8916 CKSAAP 100.00 77.65 85.50 0.7413 0.8855 Geary 100.00 77.65 85.50 0.7413 0.8855 NMBroto 100.00 77.65 85.50 0.7413 0.8855 Moran 97.83 75.29 83.21 0.6981 0.8626 CTDD 100.00 69.41 80.15 0.6659 0.8554 PAAC 95.65 75.29 82.44 0.6773 0.8518 QSOrder 93.48 72.94 80.15 0.6341 0.8288 TPC 97.83 68.24 78.63 0.6338 0.8265 GTPC 91.30 74.12 80.15 0.6247 0.8240 AAC 95.65 69.41 78.63 0.6226 0.8216 SOCNumber 93.48 65.88 75.57 0.5695 0.7987 CTDT 89.13 67.06 74.81 0.5372 0.7770 CTDC 84.78 65.88 72.52 0.4840 0.7492 GAAC 73.91 64.71 67.94 0.3688 0.6888 -
本课题组在之前的研究中,构建了预测抗体交叉或自身相互作用的集成模型CISI,数据预处理过程与本文相同,但随机分组样本不同。该模型使用的特征提取方法为三肽组分(TPC),特征选择方法为f-score,基于两个SVM集成最终的预测模型。子模型分别包含了356、346个特征,准确率分别为86.52%与89.89%,集成模型的准确率为88.20%。CISI模型中包含的特征数远远超过了样本数,易造成过拟合现象,模型泛化性低。本研究中使用不同的特征提取、特征选择方法进行改进,从特征层面与模型效能层面与CISI比较,由于CISI与本文对正负样本的定义相反,本文将敏感性定义为对有缺陷的样本的识别能力,对原文中的敏感性特异性进行转换。
如表8所示,CISI使用了TPC特征,两个子模型的特征数为300以上,而CISI2.0使用DDE特征,模型用了更少的特征数,敏感性、特异性、ACC、AUC均得到了提升,其中敏感性达到了100%,CISI2.0能识别出所有具有交叉或自身相互作用缺陷的抗体,而CISI的敏感性仅为86.05%。综上,认为相对于CISI,CISI2.0是预测抗体交叉或自身相互作用的更优模型。
表 8 CISI与CISI2.0在特征与模型层面的比较
模型 CISI CISI2.0 特征 TPC DDE 特征筛选方法 f-score MRMD2.0 每个子模型特征数 356、346 86、152 Sn/% 86.05 100 Sp/% 90.22 94.12 ACC/% 88.20 96.18 AUC 0.9609 0.9699 -
上述特征提取、特征筛选、模型构建等过程,都是使用命令或脚本完成处理的,为了方便研究人员使用本文构建的预测模型,需要开发一个用户友好的在线网页服务器。利用HTML、CSS、PHP、JavaScript、Python等语言将CISI2.0集成模型开发为在线服务工具,用户可通过以下链接访问并使用此工具:
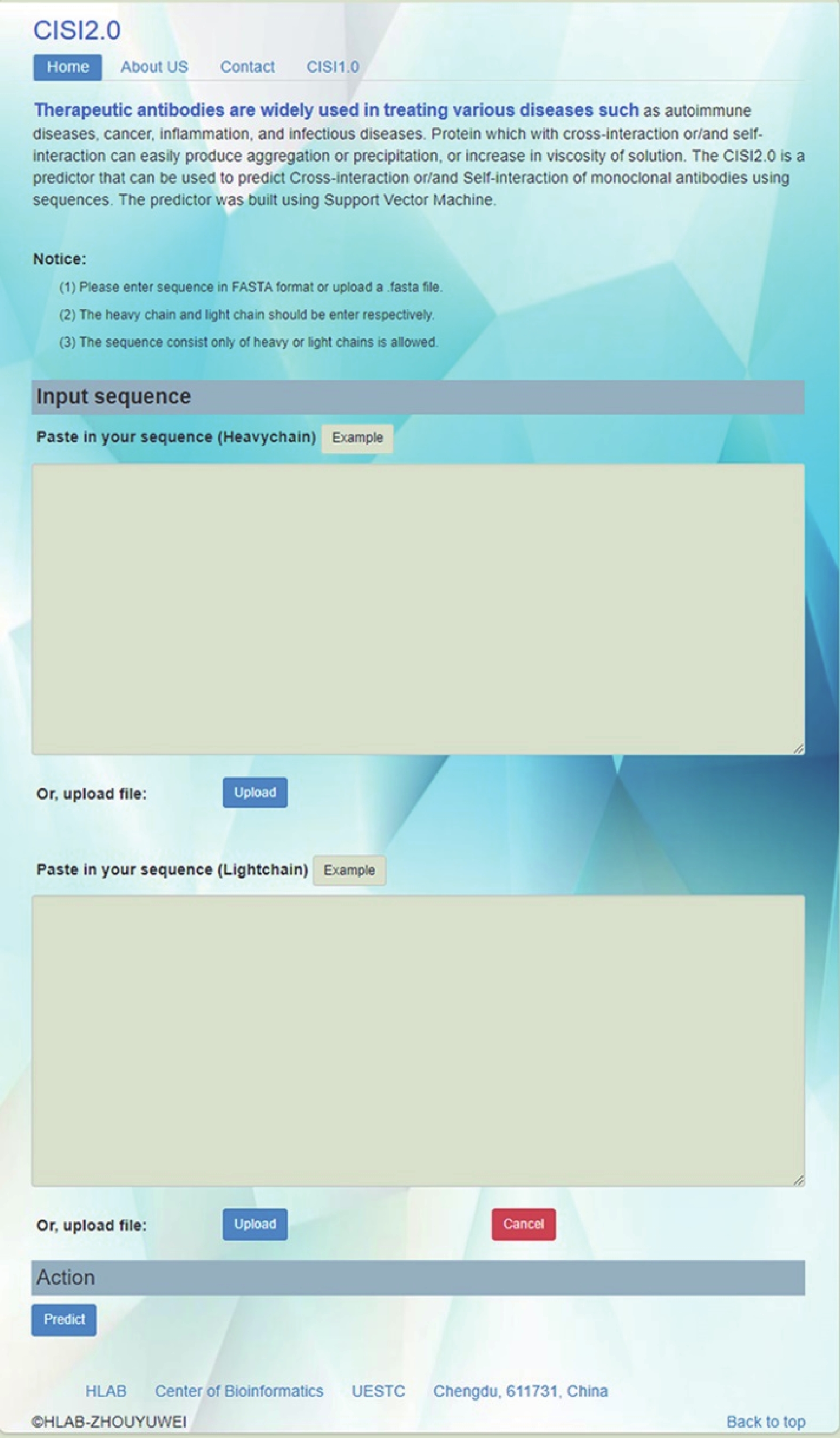
http://i.uestc.edu.cn/CISI2/ 。CISI2.0的主页如图3,要求用户分别输入FASTA格式的重链与轻链序列数据,点击“Predict”,在结果页面会返回每条抗体的预测结果,页面上方有结果说明。
图 3 CISI2.0在线工具主页面
-
进行单克隆抗体药物的可开发性评估,对治疗性抗体开发具有指导意义。在早期筛选出具有良好理化性质的先导抗体有助于提高后期开发的成功率,降低研发成本。因此,本文以临床II、III期或已批准上市的137条抗体序列与实验数据为基础,建立模型预测抗体交叉或自身相互作用。首先,根据实验测定值,将抗体序列划分为正负样本集,为了避免数据集不平衡的情况,将负样本分成与正样本数量相当的组,分别与正样本组成子数据集。然后采用DDE算法提取序列特征,MRMD2.0筛选最优特征子集,最后构建基于SVM的集成模型。最终模型的敏感性达到100%,准确率为96.18%。为了探究DDE是否为最适特征,本文与其余19种特征提取算法进行了比较,基于DDE特征构建的模型,特征数较少,并且各项评价指标均为最好,所以认为DDE能作为预测抗体交叉或自身相互作用的重要标志。除此之外,与已发表的模型相比,CISI2.0在使用较少特征的基础上,提高了模型的预测准确率,降低了过拟合风险。
当然,本文仍有一些不足:数据的规模和质量直接关系到任何一种机器学习模型的最终效能,获得可靠的数据以及正负样本的构建标准是建立模型最重要的基础。本研究中所用抗体样本量有限,无法进行独立验证。因此,需要跟进抗体数据库、相关文献的更新,以及关注抗体行业的发展,收集更多的抗体数据,使得样本含量小、样本不平衡的情况得以解决。此外,集成学习已经成为分类算法的主流。本研究的集成模型中,每个基分类器都是SVM模型,可以考虑尝试新的集成方法,同样的数据,用不同的机器学习算法构建模型,最后将不同方法的基分类器集成。
最后,希望本文提出的算法流程以及构建的预测模型能够对评估抗体可开发性相关领域提供借鉴与帮助,能作为抗体可开发性评估流程的一个环节。接下来的工作将着眼于其余影响抗体可开发性的重要因素,从结构、安全性、可制造性等,全面对抗体的可开发性进行评估。
CISI2.0: A Better Tool for Predicting Cross-Interaction or Self-Interaction of Antibodies Based on Sequences
-
摘要: 抗体广泛用于各类疾病的预防、诊断与治疗。然而,治疗性抗体研发的成功率还不尽人意。不少抗体因为稳定性差,溶解度低,存在交叉或自身相互作用等可开发性缺陷而最终开发失败。候选单克隆抗体能否开发成功,与其理化性质息息相关。虽然已有多种实验方法测定抗体交叉或自身相互作用相关的多种理化特性,但实验测试费力费时费钱。现有的抗体可开发性计算方法,或者依赖于结构,速度慢,通量低;或者未提供可用的软件或在线服务;或者提供的计算服务或软件费用过高;或者预测器的性能与健壮性有待提高。该文仅基于抗体序列,采用二肽期望均值偏差为特征,构建预测抗体交叉或自身相互作用的支持向量机模型。评估结果显示,该集成模型敏感性为100%,准确率为96.18%,可望用于抗体交叉或自身相互作用的高通量评估,加速治疗性抗体研发进程,降低研发成本。Abstract: Antibodies are widely used in the prevention, diagnosis and treatment of various diseases. However, the success rate of therapeutic antibody development is far from satisfying. Many antibodies failed due to developability problems such as poor stability, low solubility, and cross-interactions or self-interactions. Whether a candidate monoclonal antibody is developable is closely related to its physicochemical properties. Although a few experimental assays are available to detect several types of physicochemical properties of antibody relevant to cross-interactions or self-interactions, they are laborious, time-consuming and expensive. Some computational methods for antibody developability evaluation have been reported. However, these methods are slow, low throughput, not available, too expensive, or not robust enough. In this paper, a support vector machine (SVM) model for predicting cross-interaction or self-interaction of antibodies is constructed by using dipeptide deviation from expected mean derived from antibody sequences as features. The ensemble model achieves 100% sensitivity and 96.18% accuracy in cross-validation. The model can be used for high-throughput assessment of cross-interaction or self-interaction of antibodies, speeding therapeutic antibodies development, and reducing cost. Based on the model, a free web server called CISI2.0 is built, which is available at http://i.uestc.edu.cn/CISI2.
-
Key words:
- cross-interaction /
- developability /
- self-interaction /
- support vector machine /
- therapeutic antibody
-
表 1 4种评估抗体交叉或自身相互作用的实验阈值
实验 阈值 单位(标记) PSR (0–1) 0.27 None (>) AC-SINS 11.8 Nanometers (wavelength change) (>) CIC 10.1 Retention time (min) (>) CSI-BLI 0.01 BLI response units (>)  下载: 导出CSV
下载: 导出CSV
表 3 两组数据集基于留一法交叉检验的预测模型评估指标
子模型 Sn/% Sp/% ACC/% MCC AUC CISI1 76.09 69.05 72.73 0.4527 0.7257 CISI2 84.78 79.07 82.02 0.6401 0.8193
下载: 导出CSV
表 5 CISI1与CISI2共有的DDE特征
ER PY MH RW EN DR GA LW CA IN DD VA YD RA GM FS CG GG VT YI YW DK LA WT GS FM TN LH TL WF KA FN NT SI NP IY TV HW IT YF EI TM NS KK HQ
下载: 导出CSV
表 6 正样本组与负样本组的平均DDE差异比率
二肽组分 DDE比率(绝对值) TN 25.91415 FN 6.483849 GA 6.234574 IN 3.371006 MH 2.639173 YI 2.381572 TM 0.460651 IT 0.205809 DR 0.168615 WT 0.112033 NP 0.067459
下载: 导出CSV
表 7 20种特征提取算法构建的集成预测器的结果
特征 Sn/% Sp/% ACC/% MCC AUC DDE 100.00 94.12 96.18 0.9214 0.9699 DPC 100.00 82.35 88.55 0.7880 0.9157 KSCTriad 100.00 83.53 89.31 0.8002 0.9157 CKSAAGP 100.00 80.00 87.02 0.7643 0.8976 GDPC 100.00 80.00 87.02 0.7643 0.8976 CTriad 100.00 78.82 86.26 0.7527 0.8916 CKSAAP 100.00 77.65 85.50 0.7413 0.8855 Geary 100.00 77.65 85.50 0.7413 0.8855 NMBroto 100.00 77.65 85.50 0.7413 0.8855 Moran 97.83 75.29 83.21 0.6981 0.8626 CTDD 100.00 69.41 80.15 0.6659 0.8554 PAAC 95.65 75.29 82.44 0.6773 0.8518 QSOrder 93.48 72.94 80.15 0.6341 0.8288 TPC 97.83 68.24 78.63 0.6338 0.8265 GTPC 91.30 74.12 80.15 0.6247 0.8240 AAC 95.65 69.41 78.63 0.6226 0.8216 SOCNumber 93.48 65.88 75.57 0.5695 0.7987 CTDT 89.13 67.06 74.81 0.5372 0.7770 CTDC 84.78 65.88 72.52 0.4840 0.7492 GAAC 73.91 64.71 67.94 0.3688 0.6888
下载: 导出CSV
表 8 CISI与CISI2.0在特征与模型层面的比较
模型 CISI CISI2.0 特征 TPC DDE 特征筛选方法 f-score MRMD2.0 每个子模型特征数 356、346 86、152 Sn/% 86.05 100 Sp/% 90.22 94.12 ACC/% 88.20 96.18 AUC 0.9609 0.9699
下载: 导出CSV
-
[1] NORMAN R A, AMBROSETTI F, BONVIN A, et al. Computational approaches to therapeutic antibody design: Established methods and emerging trends[J]. Brief Bioinform, 2020, 21(5): 1549-1567. doi: 10.1093/bib/bbz095 [2] KAPINGIDZA A B, KOWAL K, CHRUSZCZ M. Antigen-antibody complexes[J]. Subcell Biochem, 2020, 94: 465-497. [3] NING L, ABAGNA H B, JIANG Q, et al. Development and application of therapeutic antibodies against COVID-19[J]. Int J Biol Sci, 2021, 17(6): 1486-1496. doi: 10.7150/ijbs.59149 [4] MULLARD A. FDA approves 100th monoclonal antibody product[J]. Nat Rev Drug Discov, 2021, 20(7): 491-495. doi: 10.1038/d41573-021-00079-7 [5] KAPLON H, MURALIDHARAN M, SCHNEIDER Z, et al. Antibodies to watch in 2020[J]. MAbs, 2020, 12(1): 1703531. [6] LU R M, HWANG Y C, LIU I J, et al. Development of therapeutic antibodies for the treatment of diseases[J]. J Biomed Sci, 2020, 27(1): 1. doi: 10.1186/s12929-019-0592-z [7] CARTER P J, LAZAR G A. Next generation antibody drugs: Pursuit of the 'high-hanging fruit'[J]. Nat Rev Drug Discov, 2018, 17(3): 197-223. doi: 10.1038/nrd.2017.227 [8] XU Y, WANG D, MASON B, et al. Structure, heterogeneity and developability assessment of therapeutic antibodies[J]. MAbs, 2019, 11(2): 239-264. doi: 10.1080/19420862.2018.1553476 [9] DZISOO A M, 任丽萍, 谢诗扬, 等. 治疗性抗体可开发性评估研究进展[J]. 电子科技大学学报, 2021, 50(3): 476-480. doi: 10.12178/1001-0548.2021060 DZISOO A M, REN L P, XIE S Y, et al. Progress in research on evaluation of developability of therapeutic antibody[J]. Journal of University of Electronic Science and Technology of China, 2021, 50(3): 476-480. doi: 10.12178/1001-0548.2021060 [10] JAIN T, SUN T, DURAND S, et al. Biophysical properties of the clinical-stage antibody landscape[J]. Proc Natl Acad Sci USA, 2017, 114(5): 944-949. doi: 10.1073/pnas.1616408114 [11] KRAWCZYK K, DUNBAR J, DEANE C M. Computational tools for aiding rational antibody design[J]. Methods Mol Biol, 2017, 1529: 399-416. [12] DZISOO A M, KANG J, YAO P, et al. SSH: A tool for predicting hydrophobic interaction of monoclonal antibodies using sequences[J]. Biomed Res Int, 2020, 2020: 3508107. [13] LAUER T M, AGRAWAL N J, CHENNAMSETTY N, et al. Developability index: a rapid in silico tool for the screening of antibody aggregation propensity[J]. J Pharm Sci, 2012, 101(1): 102-115. doi: 10.1002/jps.22758 [14] OBREZANOVA O, ARNELL A, CUESTA R G, et al. Aggregation risk prediction for antibodies and its application to biotherapeutic development[J]. MAbs, 2015, 7(2): 352-63. doi: 10.1080/19420862.2015.1007828 [15] DZISOO A M, HE B, KARIKARI R, et al. CISI: A tool for predicting cross-interaction or self-interaction of monoclonal antibodies using sequences[J]. Interdiscip Sci, 2019, 11(4): 691-697. doi: 10.1007/s12539-019-00330-1 [16] CHOU K C. Some remarks on protein attribute prediction and pseudo amino acid composition[J]. J Theor Biol, 2011, 273(1): 236-47. doi: 10.1016/j.jtbi.2010.12.024 [17] SARAVANAN V, GAUTHAM N. Harnessing computational biology for exact linear b-cell epitope prediction: A novel amino acid composition-based feature descriptor[J]. OMICS, 2015, 19(10): 648-658. doi: 10.1089/omi.2015.0095 [18] CARR K, MURRAY E, ARMAH E, et al. A rapid method for characterization of protein relatedness using feature vectors[J]. PLoS One, 2010, 5(3): e9550. doi: 10.1371/journal.pone.0009550 [19] CHEN Z, ZHAO P, LI F, et al. iFeature: A Python package and web server for features extraction and selection from protein and peptide sequences[J]. Bioinformatics, 2018, 34(14): 2499-2502. doi: 10.1093/bioinformatics/bty140 [20] HOSNI M, ABNANE I, IDRI A, et al. Reviewing ensemble classification methods in breast cancer[J]. Comput Methods Programs Biomed, 2019, 177: 89-112. doi: 10.1016/j.cmpb.2019.05.019 [21] SHIDA HE, FEI GUO, QUAN ZOU, et al. MRMD2.0: A python tool for machine learning with feature ranking and reduction[J]. Current Bioinformatics, 2020, 15(10): 1213-1221. [22] CORTES C, VAPNIK V. Support-vector networks[J]. Machine Learning, 1995, 20(3): 273-297. [23] KANG J, YU S, LU S, et al. Use of a 6-miRNA panel to distinguish lymphoma from reactive lymphoid hyperplasia[J]. Signal Transduct Target Ther, 2020, 5(1): 2. doi: 10.1038/s41392-019-0097-y [24] WANG Y, KANG J, LI N, et al. NeuroCS: A tool to predict cleavage sites of neuropeptide precursors[J]. Protein Pept Lett, 2020, 27(4): 337-345. doi: 10.2174/0929866526666191112150636 [25] HE B, CHEN H, HUANG J. PhD7Faster 2.0: Predicting clones propagating faster from the Ph. D. -7 phage display library by coupling PseAAC and tripeptide composition[J]. Peer J, 2019, 7: e7131. doi: 10.7717/peerj.7131 [26] CHANG C C, L C J. LIBSVM: A library for support vector machines[J]. ACM Transactions on Intelligent Systems and Technology, 2011, 2(3): 1-27. -
 点击查看大图
点击查看大图
图(3) / 表(8)
计量
- 文章访问数: 4902
- HTML全文浏览量: 1694
- PDF下载量: 77
- 被引次数: 0



















