 ISSN
ISSN 



-
随着测序成本的大幅降低,测序方法在科研和临床医疗中被广泛使用,由此产生了大量测序信息,也包括了越来越多的变异信息。基因变异导致的氨基酸变异可通过多种方式影响蛋白质的结构和功能。当变异发生在蛋白质的某些关键部位,如催化部位或配体相互作用表面,可能导致蛋白质折叠、结构不稳定或蛋白质聚集,进而导致疾病。为实现个性化医疗,追溯疾病发生的机理,预测氨基酸变异的致病性具有很高的研究价值。
与实验方法相比,计算方法具有预测成本低、效率高的突出优势。近年来,研究者们提出并不断改进了多种相关预测模型。其中,PolyPhen-2[1]面对不同预测任务灵敏性的需求,构建了HumDiv和HumVar两个数据库,运用朴素贝叶斯预测变异致病性。CADD[2]使用支持向量机算法,整合了63种基因注释,从而得到C分数来预测致病性。PON-P2[3]利用基因本体(gene ontology, GO)等特征训练,采用自抽样的方式计算置信度。DEOGEN2[4]用可视化的方式提供了每个预测的结果、相关蛋白质背景和起源。MetaSVM[5]开发了基于支持向量机的元分析框架,框架中SVM的目标函数由铰链损失和稀疏组套索组成。ClinPred[6]首次使用了ClinVar[7]数据库,并训练了两个模型,分别基于随机森林和XGBoost来获取最高预测结果。PrimateAI[8]结合6个非人类灵长类动物物种和人类的变异,共收集到38万条变异数据,训练了一个包含36层卷积神经网络的深度学习模型。
随着当前可采集的变异数据量的增加,构建一个新的融合模型以提高预测性能变得可行。因此,本文尝试使用深度学习方法从蛋白序列中提取出一些特征,将这些特征与提取并筛选的有效生物特征融合,作为模型的输入,并构建融合模型训练,旨在达到较高的预测性能。
-
本文所使用的数据包括人类、动物和植物蛋白序列中的氨基酸变异样本,如表1所示。其中,人类致病变异取自VariBench[9-10]数据库和ClinVar[7]数据库,共有17631个。按致病变异与无害变异大约1∶1的比例,从ExAC[11]数据库获取共18494个人类无害变异。对于动物变异数据:1)收集OMIA[12]数据库中有“likely causal variants”标记的变异;2)从文献[13]获得其他哺乳动物(狗、鼠和牛)的变异。动物致病氨基酸变异共317个,中性变异共312个。进一步从文献[14]取得植物变异数据集,其物种包含拟南芥、水稻和豌豆,数据集由3236个有害变异和1899个中性变异构成。
表 1 实验数据来源和分布
个 数据来源 致病变异 中性变异 总数 人类 17631 18494 36125 动物 317 312 629 植物 3236 1899 5135 总数 21184 20705 41889 由于某些蛋白序列过长,影响预测性能,本文筛选出长度不超过1500个的蛋白质序列,共9980条,包含了35179个氨基酸变异,其中致病变异18521个,无害变异16658个。
-
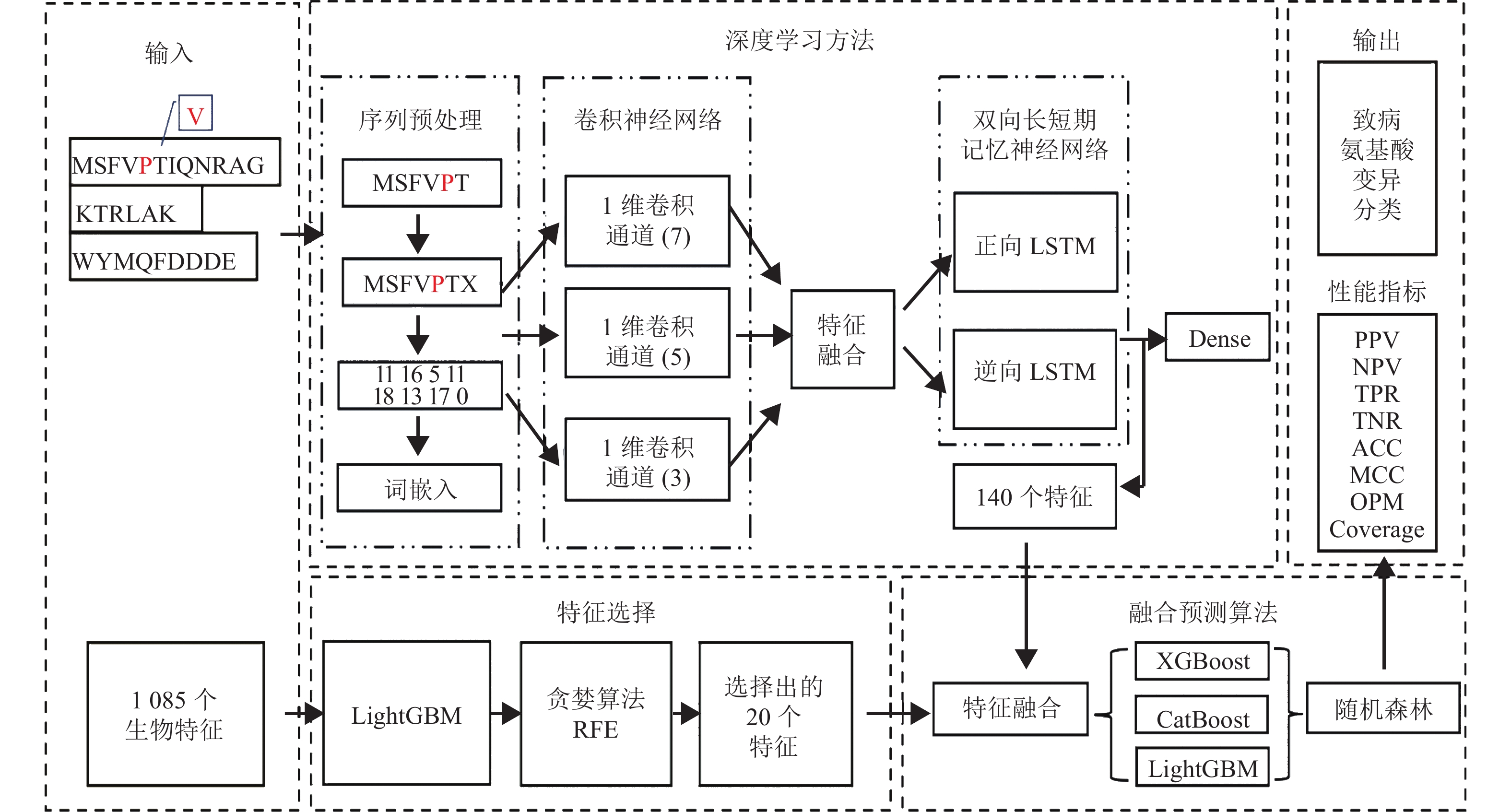
本文所构建的新型融合模型工作流程如图1所示。
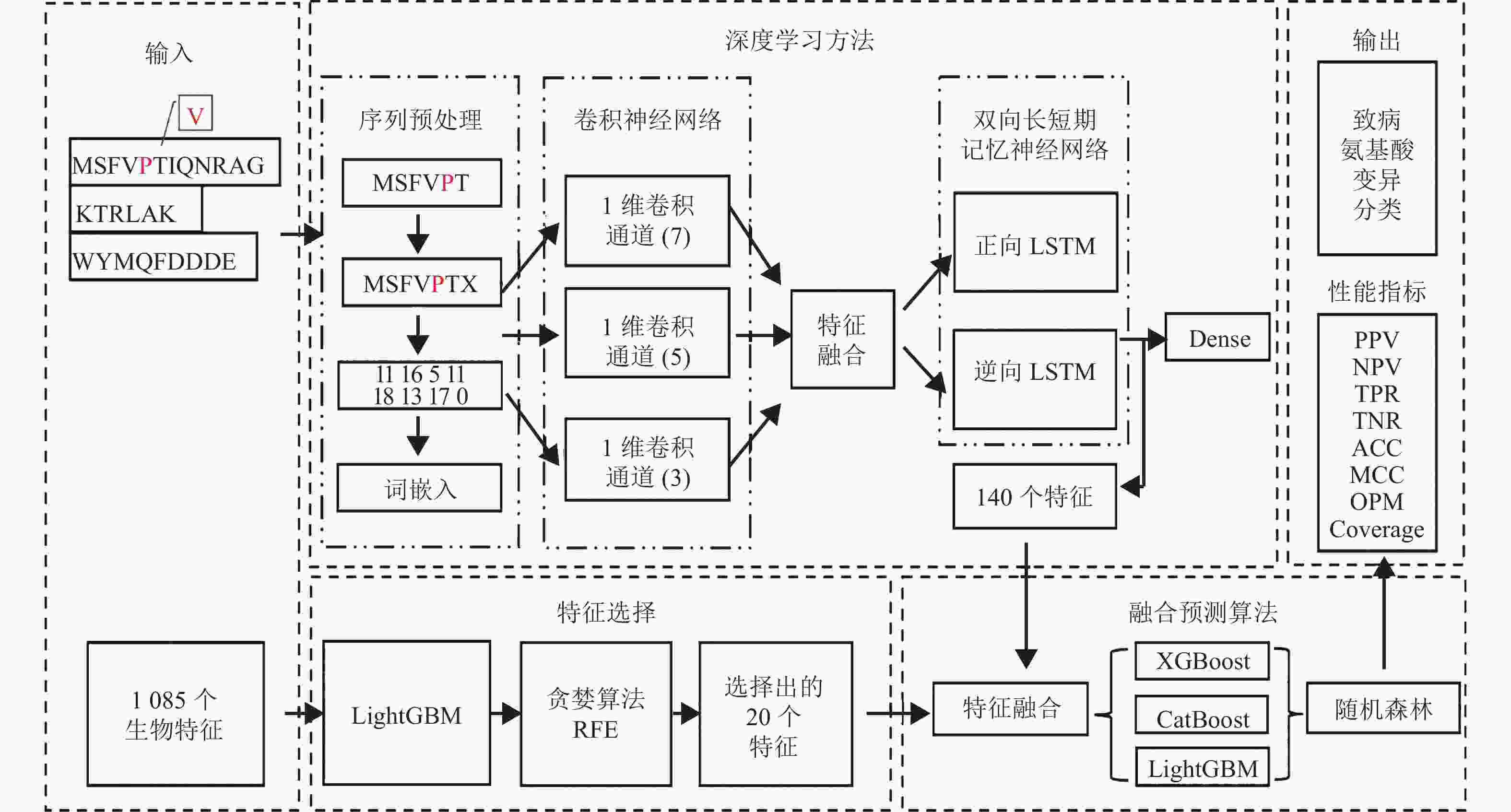
图 1 致病氨基酸变异预测的新型融合模型的工作流程
-
综合文献[3, 15]中的特征,本文共提取了1085个可能与氨基酸变异致病性有关的生物特征,包括617个氨基酸特征、436个变异类型特征、25个邻域特征、2个进化保守特征、3个蛋白质特征以及GO频率对数比特征(logical rate, LR)和功能位点特征(functional site, FS)。
617个氨基酸特征通过AAindex表[16]提取,对于每个变异,计算氨基酸变异前后得分的差异。
436个变异类型特征通过两个矩阵获得。其中,400个特征来自20×20的矩阵,矩阵的两个维度分别代表变异前和变异后的氨基酸。另外36个特征来自代表氨基酸物理和化学性质的6×6矩阵[17]。
25个邻域特征表示变异位点的序列位置特征。20维向量在23个位置,即变异位点前后11个位置的窗口中统计每种氨基酸类型的频数[18]。此外,在23个位置的邻域窗口中统计了5种氨基酸类别,包括非极性、极性、带电、带正电和带负电的频数。
使用DIAMOND[19]将每个蛋白质序列在SwissProt[20]中进行同源序列比对,找到同源序列并统计命中数。比对后,使用SIFT 4G[21]计算每个变体的SIFT分数。
3个蛋白质特征包括蛋白质序列的长度,变异是否发生在序列的第一位氨基酸,以及变异在序列中的位置。
利用朴素贝叶斯算法计算特征LR和FS如下:
1)使用R Bioconductor工具GO.db(
https://bioconductor.org/packages/GO.db/ ),从AmiGO[22]与QuickGO[23]中收集蛋白质的所有GO术语以及这些术语对应的全部上层GO术语,并过滤以避免重复。然后,在数据集中对收集到的GO术语分别统计两个类别,即致病和无害的频数,计算该蛋白GO的LR如下:$$ \mathrm{L}\mathrm{R}=\sum \mathrm{l}\mathrm{g}\frac{f\left({P}_{i}\right)+1}{f\left({N}_{i}\right)+1} $$ (1) 式中,
$ f\left({P}_{i}\right) $ 是第i个GO术语在致病集的频数;$ f\left({N}_{i}\right) $ 是第i个GO术语在无害集中的频数。为了避免分数无意义,频数都加1。如果一个蛋白质没有GO术语,则LR为0。2)根据UniProtKB/SwissProt[20]查找变异位点的Site术语,以类似LR的计算方法得到该蛋白Site的功能位点特征。
-
特征选择也称属性选择,通过从当前全部特征中筛选出部分特征的方式,达到模型某项指标最优化的目的。较多的特征中可能存在无关特征和冗余特征,这也许会导致维度灾难,使得模型的性能下降,训练的时间开销增加。当特征较多而样本数量较少时,模型往往容易出现过拟合的问题。为了避免特征冗余和模型过拟合的风险,减少算力资源浪费,增加模型可解释性,需要筛选出与分类性能密切相关的生物特征。
参照文献[1, 3]的特征选择方法,本文使用LightGBM算法结合递归特征消除(recursive feature elimination, RFE)方法[24]进行特征选择。特征选择的过程是以全部特征构建模型,为每个特征分配权重,并去除权重最小的特征,然后在剩余的特征上重复上述操作,直到达到预设的特征数量。
-
随着数据量增加,有望通过深度学习方法提取出一些没有明确生物意义但对分类有帮助的特征。本文构建的深度学习网络主要包括两个部分:卷积神经网络(convolutional neural networks, CNN)和双向长短期记忆神经网络(bi-directional long-short term memory, Bi-LSTM)。
CNN提取特征的基本思路是通过不断平移卷积核,对原始矩阵的每个元素进行卷积操作,从而提取到某些细节特征。卷积核的感受野与卷积核大小有关,感受野不同,卷积网络能提取到的特征细节也不一样。本模型设置了多条卷积线路,不同线路对应不同卷积核。通过融合多种卷积核的卷积结果,可以提取到较全面的特征。本模块使用了多层的小卷积核,代替少层的大卷积核。在相同大小感受野的前提下,能够缩小计算量,提高训练效率。对于每层卷积提取的特征,再利用激活函数进行非线性变换,以提取更具有泛化性的特征。
特征提取中,本文进一步使用了Bi-LSTM,其构造中有记忆门和遗忘门,经过训练,能够选择记忆和遗忘某些信息,探索到CNN输出的特征向量中距离较远的关系。Bi-LSTM由正向LSTM和逆向LSTM组成,氨基酸变异位点特征同时与该位点前后序列都有关。经过特征累计,Bi-LSTM能够提取出位点前后向的双向特征。
-
提取并筛选出的生物特征与深度学习提取到的特征以拼接的形式相融合,作为融合预测算法的输入。融合特征与预测结果之间的关系比较复杂,多个模型的融合或许能取得较好的性能。不同的模型从不同的角度去观测数据。堆叠法(stacking)可以整合多个模型观测数据的角度,相互取长补短,能够得到一个更加全面和优秀的结果。LightGBM[25]采用了基于梯度的单边采样,留下梯度大的样本,随机抽样梯度小的样本,来分割数据样本。XGBoost[26]通过直方图算法,将全部样本按照某个特征分成离散区域,并根据这些区域确定最佳分割。CatBoost采用贪婪策略,整合不同类别型特征得到新的特征,能够直接处理类别型特征。所以,XGBoost、CatBoost和LightGBM这3种提升boosting算法各有特点。
本融合模型选择这3个算法作为第一层模型,训练并保留预测结果,得到3列新的特征表达。第二层随机森林用得到的特征表达进行训练,得到最终的分类结果。以此达到平衡各算法的优缺点,提高分类性能的目的。
-
本文引入了一些评价指标对模型进行评估,来直观辨别分类性能的优劣,其中包括阳性预测值(PPV)、阴性预测值(NPV)、敏感性(TPR)、特异性(TNR)、准确性(ACC)、马修斯相关系数(MCC)和总体绩效指标(overall performance measure, OPM)。这些评价指标的数学定义如下:
$$ \mathrm{P}\mathrm{P}\mathrm{V}=\frac{\mathrm{T}\mathrm{P}}{\mathrm{T}\mathrm{P}+\mathrm{F}\mathrm{P}} $$ (2) $$ \mathrm{N}\mathrm{P}\mathrm{V}=\frac{\mathrm{T}\mathrm{N}}{\mathrm{T}\mathrm{N}+\mathrm{F}\mathrm{N}} $$ (3) $$ \mathrm{T}\mathrm{P}\mathrm{R}=\frac{\mathrm{T}\mathrm{P}}{\mathrm{T}\mathrm{P}+\mathrm{F}\mathrm{N}} $$ (4) $$ \mathrm{T}\mathrm{N}\mathrm{R}=\frac{\mathrm{T}\mathrm{N}}{\mathrm{T}\mathrm{N}+\mathrm{F}\mathrm{P}} $$ (5) $$ \mathrm{A}\mathrm{C}\mathrm{C}=\frac{\mathrm{T}\mathrm{P}+\mathrm{T}\mathrm{N}}{\mathrm{T}\mathrm{P}+\mathrm{T}\mathrm{N}+\mathrm{F}\mathrm{P}+\mathrm{F}\mathrm{N}} $$ (6) $$ {\rm{MCC}} = \frac{{{\rm{TP}} \times {\rm{TN}} - {\rm{FP}} \times {\rm{FN}}}}{{\sqrt {({\rm{TP}} + {\rm{FN}})({\rm{TP}} + {\rm{FP}})({\rm{TN}} + {\rm{FN}})({\rm{TN}} + {\rm{FP}})} }} $$ (7) $$ \mathrm{n}\mathrm{M}\mathrm{C}\mathrm{C}=\frac{\mathrm{M}\mathrm{C}\mathrm{C}+1}{2} $$ (8) $$ \mathrm{O}\mathrm{P}\mathrm{M}=\frac{(\mathrm{P}\mathrm{P}\mathrm{V}+\mathrm{N}\mathrm{P}\mathrm{V})(\mathrm{T}\mathrm{P}\mathrm{R}+\mathrm{T}\mathrm{N}\mathrm{R})(\mathrm{A}\mathrm{C}\mathrm{C}+\mathrm{n}\mathrm{M}\mathrm{C}\mathrm{C})}{8} $$ (9) MCC在TP、TN、FP和FN基础上评价二元分类性能,其值在−1~+1之间,值越大分类性能越好。OPM由PON-P2[3]定义,将MCC归一化为0~1值(nMCC),结合PPV、NPV、TPR、TNR和ACC,如式(9),两两之和为棱长作长方体,长方体体积的1/8作为OPM的值。OPM的取值范围为0~1,值越接近1,分类性能越好。
-
在数据集中随机选取90%的变异样本作为训练集进行10重交叉验证。实验用的部分数据和代码已经上传GitHub (
https://github.com/s1mpleCN/PON-DML )。 -
参照文献[1, 3]的特征筛选经验,本文用RFE算法分别筛选了100、50、20、10个特征进行比较。其交叉验证结果如表2所示,其中仅基于20个生物特征,预测准确性ACC即可达到89.7%,MCC为79.4%,OPM为72.2%。综合考虑特征数量和模型性能的平衡,本文最终选择了筛选出的20个生物特征来进行特征融合。
表 2 筛选不同数量特征的CV结果
评价指标 10个
特征20个
特征50个
特征100个
特征全部
特征阳性预测值(PPV) 0.910 0.909 0.909 0.908 0.908 阴性预测值(NPV) 0.882 0.885 0.885 0.884 0.885 敏感性(TPR) 0.892 0.896 0.896 0.895 0.895 特异性(TNR) 0.901 0.899 0.899 0.899 0.898 准确性(ACC) 0.896 0.897 0.898 0.897 0.897 马修斯相关系数(MCC) 0.792 0.794 0.795 0.793 0.793 总体绩效指标(OPM) 0.719 0.722 0.723 0.720 0.721 -
考虑到原始蛋白序列是由英文字母组成的,无法直接用深度学习方法训练。借鉴自然语言处理的做法,将原始蛋白质序列按照氨基酸单字母缩写的顺序从小到大进行编码[27]。神经网络中的Bi-LSTM要求输入的序列长度限制为固定长度,在长度短的序列后面填充0,使序列最终长度均为1500个氨基酸。通过词嵌入方法将序列中每个数字编码,转化为一个实数向量。本文设置嵌入参数为128,输出为1500×128的矩阵。
本文构建的CNN使用了一维卷积函数,只需要定义核大小的一个参数,另一参数默认取词嵌入部分设置的长度128。CNN共构造了3条卷积路线,分别对应3种大小的卷积核(7×128、5×128和3×128)。每条路线中都包含5个卷积层,每个卷积层之后都进行最大池化操作,从而提取出更显著的特征。将3条路线提取的特征以拼接的方式融合,作为Bi-LSTM的输入。经过实验比较,Bi-LSTM层的输出参数设为70时,预测性能最好。此时,正向LSTM和逆向LSTM都可以提取到70个特征。
在构建的神经网络中加入丢弃层(dropout),每次随机选择训练过程中的部分神经元进行计算,避免模型过拟合。网络中加入批标准化层(batch normalization),可以避免梯度爆炸,加快迭代速度。综合考虑模型的准确性和泛化性,利用早停机制(early stopping),共进行30次迭代。深度学习网络参数详见表3。
表 3 深度学习网络中的参数
网络层 参数 输入 sentence_length=1500; n_batches=512 丢弃层 rate=0.5 嵌入层 input_dim=21; output_dim=128 卷积网络层 5层filters=32,32,64,64,128;
3种filter_length=(75或3); activation=relu最大池化层 default 批标准化层 default 双向长短期记忆神经网络层 lstm_output_size=70; dropout=0.2 全连接层 units=8; activation=relu 丢弃层 rate=0.2 全连接层 units=1; activation=sigmoid 神经网络训练的模型性能如表4所示,其准确性ACC为84.9%,MCC为69.7%,OPM达到61.2%。
表 4 神经网络训练的模型性能结果
评价指标 神经网络模型 阳性预测值(PPV) 0.844 阴性预测值(NPV) 0.855 敏感性(TPR) 0.875 特异性(TNR) 0.821 准确性(ACC) 0.849 马修斯相关系数(MCC) 0.697 总体绩效指标(OPM) 0.612 -
如图2所示,本融合模型将每次交叉验证的训练集随机分成3份,分两层进行训练。首先选择XGBoost,每一次用其中两份数据预测剩下的1份数据以及验证集的数据,保留得到的预测结果。每份数据都预测一次,共可以保留3份训练集的预测结果和3份验证集的预测结果。整合训练集的预测结果作为第二层的一列新的特征表达。将3份验证集的预测结果取平均值用作第二层验证集的一列特征表达。然后用CatBoost和LightGBM重复上面步骤。最后,在第二层中用随机森林进行训练预测,相当于对第一层的模型做了融合。
本文融合模型交叉验证结果如表5所示,其中准确性ACC为92.8%,MCC达到85.5%,OPM值为79.8%。
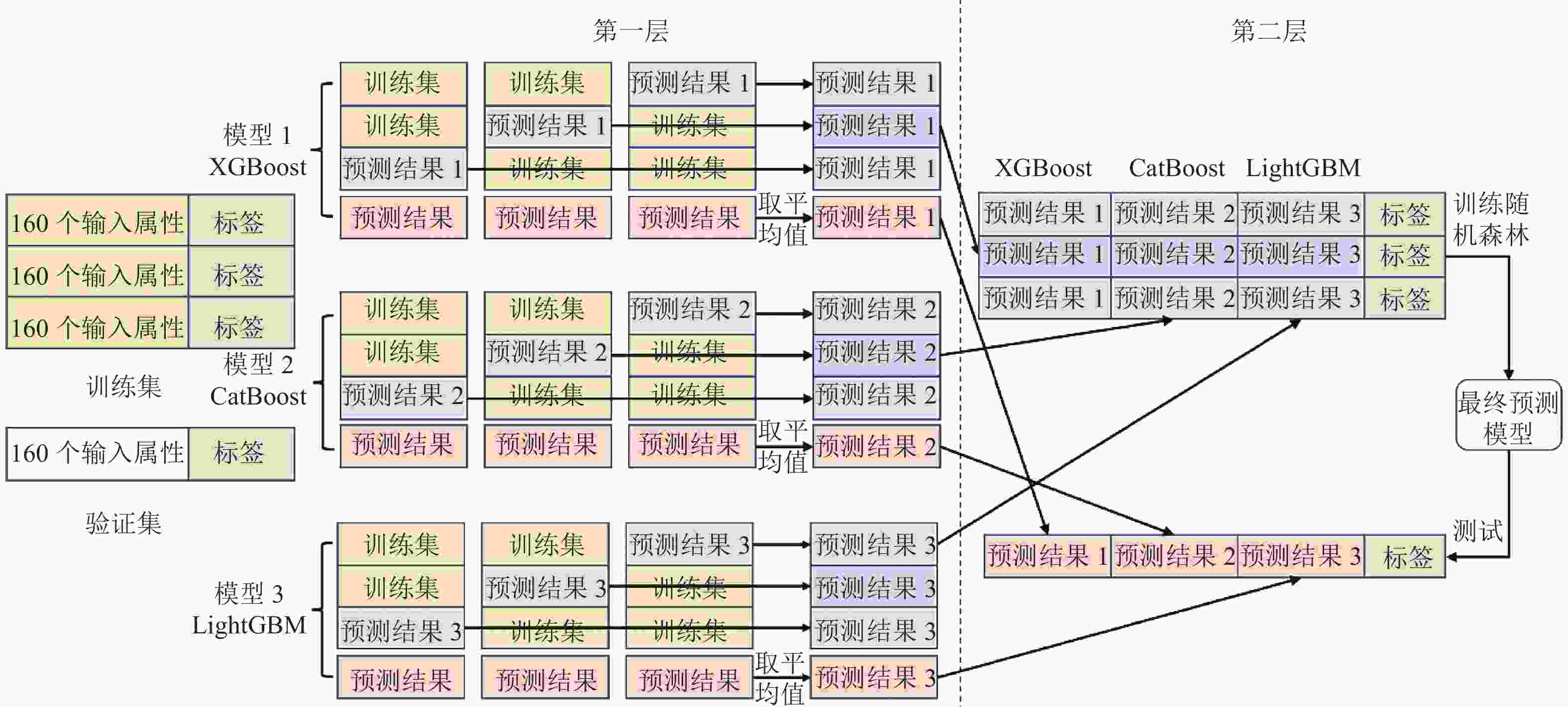
图 2 融合预测算法工作流程
表 5 融合模型10重交叉验证结果
评价指标 本文融合模型 阳性预测值(PPV) 0.932 阴性预测值(NPV) 0.922 敏感性(TPR) 0.930 特异性(TNR) 0.925 准确性(ACC) 0.928 马修斯相关系数(MCC) 0.855 总体绩效指标(OPM) 0.798 -
以剩余的10%数据作为盲测集,使用构建好的融合模型进行预测。其结果如表6第二列所示,本文融合模型准确性ACC达到93.1%,MCC为86.1%,OPM为80.6%。
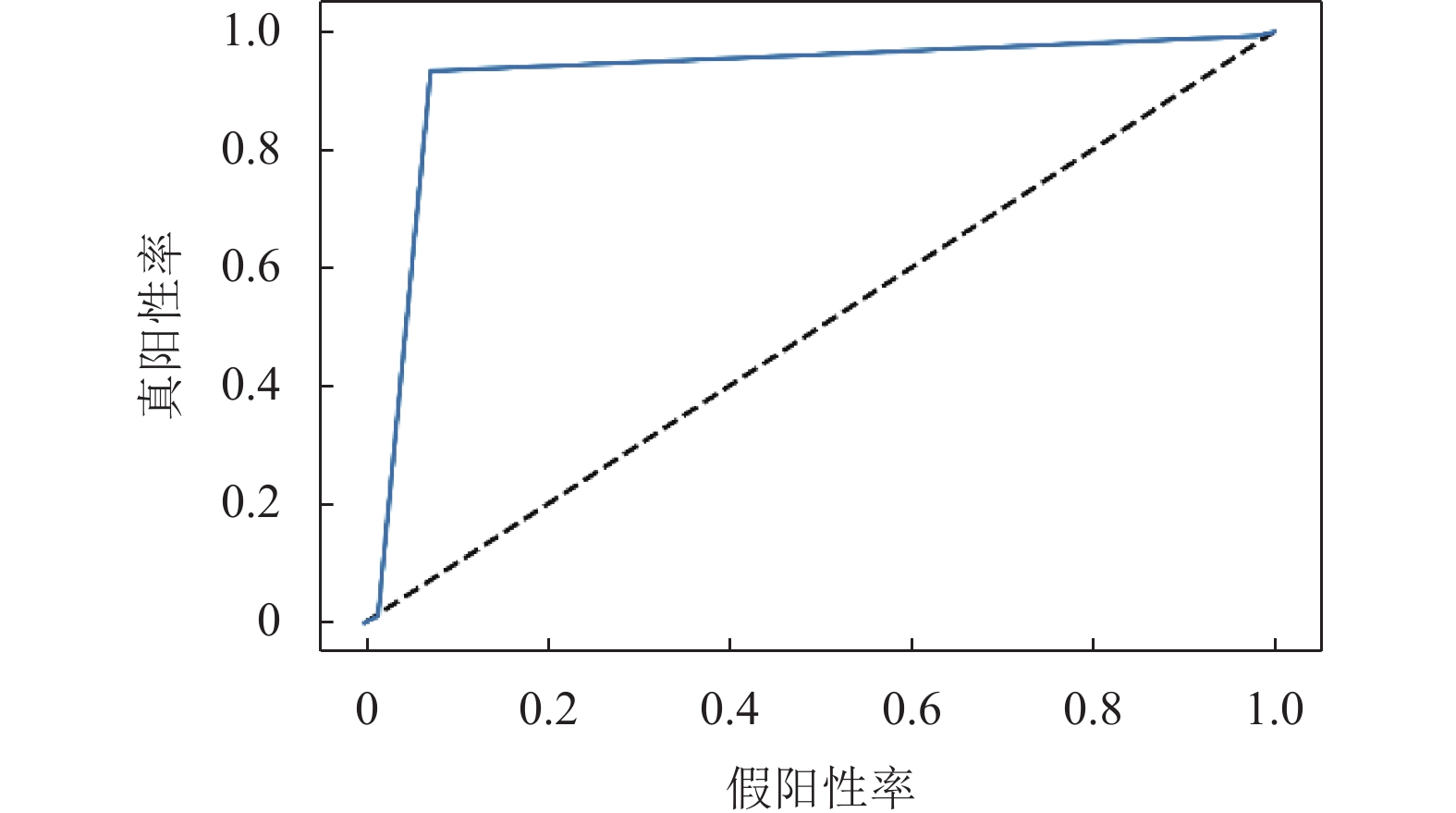
本文融合模型在盲测集上的ROC曲线如图3所示,曲线下面积(AUC)为0.921。
本文还在该盲测集上比较了几种常见的预测工具性能,包括Polyphen2[1]、CADD[2]、PON-P2[3]、DEOGEN2[4]、MetaSVM[5]、ClinPred[6]和PrimateAI[8]。其中CADD阈值设为20时,其性能取得最佳。
由表6可见,Polyphen2准确性较低,为75.0%。考虑到Polyphen2训练的数据量仅为2万条,限制了它的性能。CADD准确性达到77.5%。该模型以36个其他预测模型的得分作为特征,或许某些预测模型并不能有效提高氨基酸变异致病性的预测性能。定义覆盖率为模型能够预测的变异数/测试集总样本数量。PON-P2准确性最高,达到93.7%,但覆盖率仅为54.2%,这是因为PON-P2舍弃了置信度不高的变异预测。而本融合模型覆盖率达到了100.0%。由于综合了两个预测模型,ClinPred准确性较高,为91.1%,但覆盖率也仅为76.5%。PrimateAI是一个基于深度学习的预测模型,直接从序列中提取特征,准确性较低,仅为56.0%。可见生物特征在变异致病性预测中扮演着更为重要的角色。
表 6 与常用的预测模型在盲测集上的性能结果比较
评价指标 本文融合模型 Polyphen2 CADD PON-P2 DEOGEN2 MetaSVM ClinPred PrimateAI 阳性预测值(PPV) 0.932 0.702 0.716 0.962 0.945 0.950 0.968 0.827 阴性预测值(NPV) 0.930 0.832 0.877 0.914 0.843 0.845 0.866 0.556 敏感性(TPR) 0.934 0.877 0.909 0.908 0.822 0.825 0.850 0.264 特异性(TNR) 0.927 0.621 0.643 0.964 0.952 0.957 0.972 0.943 准确性(ACC) 0.931 0.750 0.775 0.937 0.887 0.891 0.911 0.600 马修斯相关系数(MCC) 0.861 0.516 0.572 0.874 0.780 0.788 0.828 0.282 总体绩效指标(OPM) 0.806 0.433 0.483 0.822 0.704 0.713 0.762 0.259 覆盖率(Coverage) 1.000 0.755 0.769 0.542 0.766 0.765 0.765 0.744 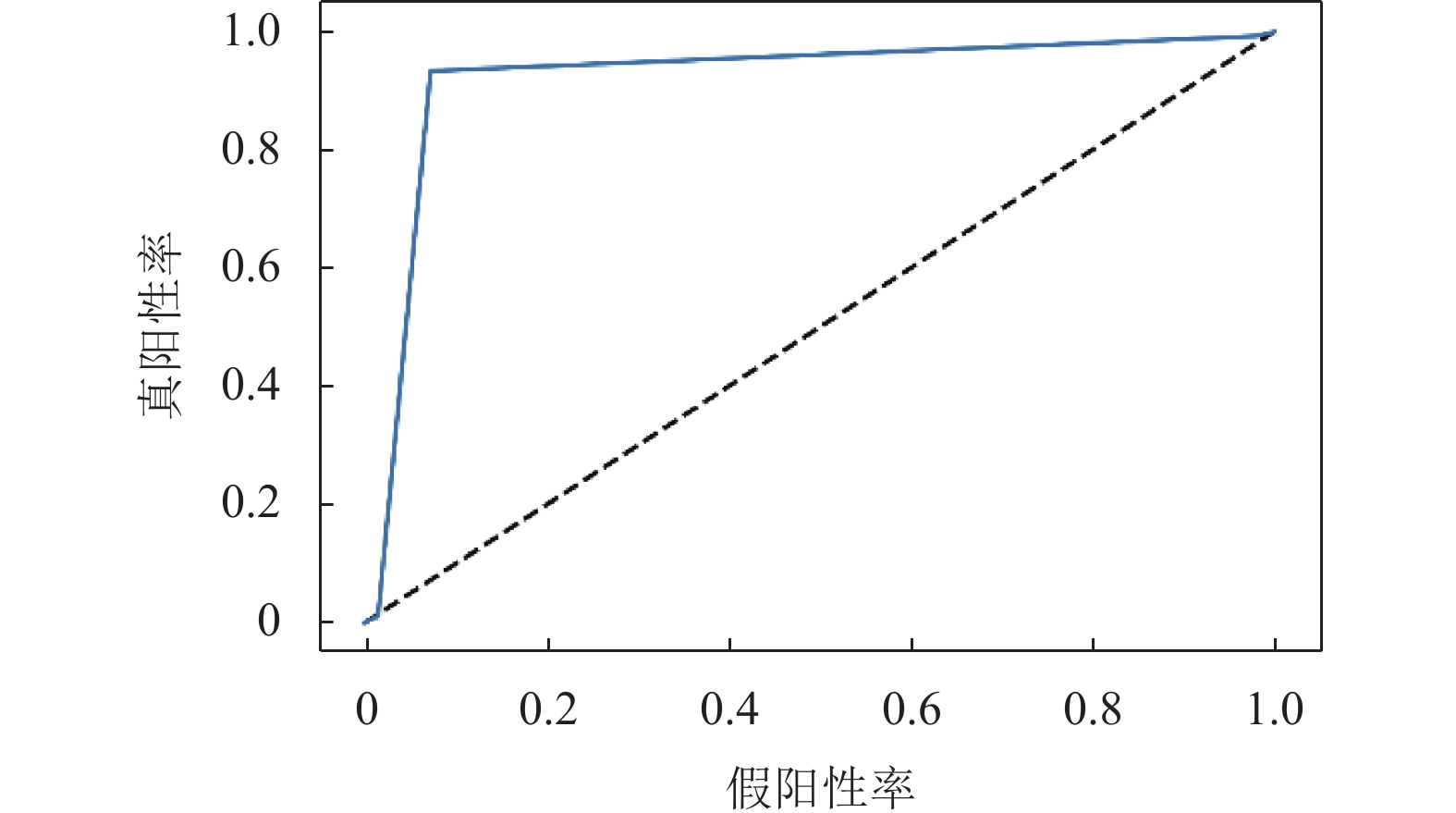
图 3 盲测集ROC曲线
-
氨基酸变异常常会对蛋白质的结构和功能造成影响,进而导致疾病。基于计算的方法作为一种预测氨基酸变异致病性的有效途径,被研究者广泛使用。为了提高预测准确性和泛化性,本文构建了一个新型融合模型。首先,收集到包含人类、动物和植物种群的氨基酸变异作为数据集。接着,提取有效生物特征,并用RFE筛选出最优特征子集。然后,使用深度学习网络提取特征,深度学习网络由CNN和Bi-LSTM组成。将筛选完的生物特征和深度学习网络提取的特征以拼接的方式融合,作为预测输入。最后,构建一个基于XGBoost、CatBoost、LightGBM和随机森林的融合模型,得到最终的预测结果。融合模型的交叉验证准确性ACC达到92.8%,MCC达到85.5%,OPM值为79.8%。与其他工具相比,本文模型具有更高的准确性和泛化性。该模型工具可用于辅助临床诊断和药物设计,降低研发成本。
A Novel Fusion Model for Predicting Pathogenic Amino Acid Substitution
-
摘要: 氨基酸变异常常会影响蛋白质的结构和功能,进而导致疾病。当前,研究者们已经提出了一些基于计算的方法来预测氨基酸变异致病性。该文构建了一个新型融合模型,旨在提高预测性能和泛化性。首先,提取影响致病性的各类生物特征并用递归特征消除RFE方法筛选最优特征子集。然后,建立包含卷积神经网络和双向长短期记忆神经网络的深度学习模型提取特征,并以拼接的方式融合这两类特征作为模型输入。最后,构建一个基于XGBoost、CatBoost、LightGBM和随机森林的融合模型,用以预测氨基酸变异致病性。该融合模型的10重交叉验证准确性为92.8%,盲测准确性为93.1%,取得了当前最高的预测准确性和泛化性。该工具可用于辅助临床诊断和药物设计,降低研发成本。
-
关键词:
- 氨基酸变异 /
- 双向长短期记忆神经网络 /
- 卷积神经网络 /
- 融合模型 /
- 致病性预测
Abstract: Amino acid substitution often affects the structure and function of proteins, leading to diseases. At present, researchers have proposed some computational methods to predict the pathogenicity of amino acid substitution. This paper constructs a new fusion model to improve the prediction performance and generalization. Firstly, various biological features affecting pathogenicity are extracted and the optimal feature subset is screened by recursive feature elimination (RFE) method. Then, a deep learning model including convolutional neural networks and bi-directional long-short term memory is established to extract features, and the two types of features are fused in a splicing way as model input. Finally, a fusion model based on XGBoost, CatBoost, LightGBM and Random Forest is constructed to predict the pathogenicity of amino acid substitution. The 10-fold cross validation accuracy of the fusion model is 92.8%, and the blind test accuracy is 93.1%, achieving the highest prediction accuracy and generalization to date. The tool can be used to assist clinical diagnosis and drug design and reduce research and development costs. -
表 1 实验数据来源和分布
个 数据来源 致病变异 中性变异 总数 人类 17631 18494 36125 动物 317 312 629 植物 3236 1899 5135 总数 21184 20705 41889  下载: 导出CSV
下载: 导出CSV
表 2 筛选不同数量特征的CV结果
评价指标 10个
特征20个
特征50个
特征100个
特征全部
特征阳性预测值(PPV) 0.910 0.909 0.909 0.908 0.908 阴性预测值(NPV) 0.882 0.885 0.885 0.884 0.885 敏感性(TPR) 0.892 0.896 0.896 0.895 0.895 特异性(TNR) 0.901 0.899 0.899 0.899 0.898 准确性(ACC) 0.896 0.897 0.898 0.897 0.897 马修斯相关系数(MCC) 0.792 0.794 0.795 0.793 0.793 总体绩效指标(OPM) 0.719 0.722 0.723 0.720 0.721
下载: 导出CSV
表 3 深度学习网络中的参数
网络层 参数 输入 sentence_length=1500; n_batches=512 丢弃层 rate=0.5 嵌入层 input_dim=21; output_dim=128 卷积网络层 5层filters=32,32,64,64,128;
3种filter_length=(75或3); activation=relu最大池化层 default 批标准化层 default 双向长短期记忆神经网络层 lstm_output_size=70; dropout=0.2 全连接层 units=8; activation=relu 丢弃层 rate=0.2 全连接层 units=1; activation=sigmoid
下载: 导出CSV
表 4 神经网络训练的模型性能结果
评价指标 神经网络模型 阳性预测值(PPV) 0.844 阴性预测值(NPV) 0.855 敏感性(TPR) 0.875 特异性(TNR) 0.821 准确性(ACC) 0.849 马修斯相关系数(MCC) 0.697 总体绩效指标(OPM) 0.612
下载: 导出CSV
表 5 融合模型10重交叉验证结果
评价指标 本文融合模型 阳性预测值(PPV) 0.932 阴性预测值(NPV) 0.922 敏感性(TPR) 0.930 特异性(TNR) 0.925 准确性(ACC) 0.928 马修斯相关系数(MCC) 0.855 总体绩效指标(OPM) 0.798
下载: 导出CSV
表 6 与常用的预测模型在盲测集上的性能结果比较
评价指标 本文融合模型 Polyphen2 CADD PON-P2 DEOGEN2 MetaSVM ClinPred PrimateAI 阳性预测值(PPV) 0.932 0.702 0.716 0.962 0.945 0.950 0.968 0.827 阴性预测值(NPV) 0.930 0.832 0.877 0.914 0.843 0.845 0.866 0.556 敏感性(TPR) 0.934 0.877 0.909 0.908 0.822 0.825 0.850 0.264 特异性(TNR) 0.927 0.621 0.643 0.964 0.952 0.957 0.972 0.943 准确性(ACC) 0.931 0.750 0.775 0.937 0.887 0.891 0.911 0.600 马修斯相关系数(MCC) 0.861 0.516 0.572 0.874 0.780 0.788 0.828 0.282 总体绩效指标(OPM) 0.806 0.433 0.483 0.822 0.704 0.713 0.762 0.259 覆盖率(Coverage) 1.000 0.755 0.769 0.542 0.766 0.765 0.765 0.744
下载: 导出CSV
-
[1] ADZHUBEI I A, SCHMIDT S, PESHKIN L, et al. A method and server for predicting damaging missense mutations[J]. Nature Methods, 2010, 7(4): 248-249. doi: 10.1038/nmeth0410-248 [2] RENTZSCH P, WITTEN D, COOPER G M, et al. CADD: Predicting the deleteriousness of variants throughout the human genome[J]. Nuclc Acids Research, 2019, 47(D1): 886-894. doi: 10.1093/nar/gky1016 [3] NIROULA A, UROLAGIN S, VIHINEN M. PON-P2: Prediction method for fast and reliable identification of harmful variants[J]. Plos One, 2015, 10(2): e0117380. doi: 10.1371/journal.pone.0117380 [4] RAIMONDI D, TANYALCIN I, FERTÉ J, et al. DEOGEN2: Prediction and interactive visualization of single amino acid variant deleteriousness in human proteins[J]. Nucleic Acids Research, 2017, 45(W1): 201-206. doi: 10.1093/nar/gkx390 [5] KIM S, JHONG J H, LEE J J, et al. Meta-analytic support vector machine for integrating multiple omics data[J]. BioData Mining, 2017, 10(1): 1-14. doi: 10.1186/s13040-016-0121-5 [6] ALIREZAIE N, KERNOHAN K D, HARTLEY T, et al. ClinPred: Prediction tool to identify disease-relevant nonsynonymous single-nucleotide variants[J]. The American Journal of Human Genetics, 2018, 103(4): 474-483. doi: 10.1016/j.ajhg.2018.08.005 [7] LANDRUM M J, LEE J M, RILEY G R, et al. ClinVar: Public archive of relationships among sequence variation and human phenotype[J]. Nucleic Acids Research, 2014, 42(D1): 980-985. doi: 10.1093/nar/gkt1113 [8] SUNDARAM L, GAO H, PADIGEPATI S R, et al. Predicting the clinical impact of human mutation with deep neural networks[J]. Nature Genetics, 2018, 50(8): 1161-1170. doi: 10.1038/s41588-018-0167-z [9] SARKAR A, YANG Y, VIHINEN M. Variation benchmark datasets: Update, criteria, Quality and applications[J]. Database, 2020, DOI: 10.1093/database/baz117. [10] NAIR P S, VIHINEN M. VariBench: A benchmark database for variations[J]. Human Mutation, 2013, 34(1): 42-49. doi: 10.1002/humu.22204 [11] KARCZEWSKI K J, WEISBURD B, THOMAS B, et al. The ExAC browser: Displaying reference data information from over 60 000 exomes[J]. Nucleic Acids Research, 2017, 45(D1): 840-845. doi: 10.1093/nar/gkw971 [12] NICHOLAS F W. Online mendelian inheritance in animals (OMIA): A comparative knowledgebase of genetic disorders and other familial traits in non-laboratory animals[J]. Nucleic Acids Research, 2003, 31(1): 275-277. doi: 10.1093/nar/gkg074 [13] PLEKHANOVA E, NUZHDIN S V, UTKIN L V, et al. Prediction of deleterious mutations in coding regions of mammals with transfer learning[J]. Evolutionary Applications, 2019, 12(1): 18-28. doi: 10.1111/eva.12607 [14] KOVALEV M S, IGOLKINA A A, SAMSONOVA M G, et al. A pipeline for classifying deleterious coding mutations in agricultural plants[J]. Frontiers in Plant Science, 2018, 9: 1734. doi: 10.3389/fpls.2018.01734 [15] YANG Y, NIROULA A, SHEN B R, et al. PON-Sol: Prediction of effects of amino acid substitutions on protein solubility[J]. Bioinformatics, 2016, 32(13): 2032-2034. doi: 10.1093/bioinformatics/btw066 [16] KAWASHIMA S, KANEHISA M. AAindex: Amino acid index database[J]. Nucleic Acids Research, 2000, 28(1): 374. doi: 10.1093/nar/28.1.374 [17] SHEN B R, VIHINEN M. Conservation and covariance in PH domain sequences: Physicochemical profile and information theoretical analysis of XLA-causing mutations in the Btk PH domain[J]. Protein Engineering Design and Selection, 2004, 17(3): 267-276. doi: 10.1093/protein/gzh030 [18] LOCKWOOD S, KRISHNAMOORTHY B, YE P. Neighborhood properties are important determinants of temperature sensitive mutations[J]. Plos One, 2011, 6(12): e28507. doi: 10.1371/journal.pone.0028507 [19] BUCHFINK B, XIE C, HUSON D H. Fast and sensitive protein alignment using DIAMOND[J]. Nature Methods, 2015, 12(1): 59-60. doi: 10.1038/nmeth.3176 [20] SHOMER B. Seqalert-a daily sequence alertness server for the Embl and Swissprot databases[J]. Bioinformatics, 1997, 13(5): 545-547. doi: 10.1093/bioinformatics/13.5.545 [21] VASER R, ADUSUMALLI S, LENG S N, et al. SIFT missense predictions for genomes[J]. Nature Protoc, 2016, 11(1): 1-9. doi: 10.1038/nprot.2015.123 [22] CARBON S, IRELAND A, MUNGALL C J, et al. AmiGO: Online access to ontology and annotation data[J]. Bioinformatics, 2009, 25(2): 288-289. doi: 10.1093/bioinformatics/btn615 [23] MUNOZ-TORRES M, CARBON S. Get GO! Retrie ving GO data using AmiGO, QuickGO, API, files, and tools[M]. New York: Springer, 2017. [24] GUYON I, WESTON J, BARNHILL S, et al. Gene selection for cancer classification using support vector machines[J]. Machine Learning, 2002, 46(1): 389-422. [25] KE G, MENG Q, FINLEY T, et al. Lightgbm: A highly efficient gradient boosting decision tree[J]. Advances in Neural Information Processing Systems, 2017, 30: 3146-3154. [26] CHEN T, GUESTRIN C. Xgboost: A scalable tree boosting system[C]//Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. San Francisco: Association for Computing Machinery, 2016: 785-794. [27] 李洪顺, 于华, 宫秀军. 一种只利用序列信息预测RNA结合蛋白的深度学习模型[J]. 计算机研究与发展, 2018, 55(1): 93-101. doi: 10.7544/issn1000-1239.2018.20160508 LI H S, YU H, GONG X J. A deep learning model for predicting RNA-binding proteins only from primary sequences[J]. Journal of Computer Research and Development, 2018, 55(1): 93-101. doi: 10.7544/issn1000-1239.2018.20160508 -
 点击查看大图
点击查看大图
图(3) / 表(6)
计量
- 文章访问数: 4054
- HTML全文浏览量: 1217
- PDF下载量: 41
- 被引次数: 0











