 ISSN
ISSN 


 下载:
下载:
-
多序列比对是生物信息学研究中重要的课题之一,对于识别未知基因功能、分析物种间的进化关系、识别基因之间的保守区域等问题有着重要作用。随着测序技术的发展,基因序列数据快速增长,现有软件难以处理大规模的多序列比对问题。
目前大多数软件采用的是渐进式比对策略或者迭代式比对策略[1],如MAFFT[2]、Kalign3[3]、Clustal[4-5]、MUSCLE[6]、T-Coffee[7]、HAlign[8]等。渐进式比对需要先计算两两序列之间的距离,再根据距离矩阵使用层次聚类算法,如UPGMA、Neighbor Joining等构建一颗比对的指导树,沿着指导树的枝干进行两两比对与合并,最后得到最终结果。而迭代式比对策略在此基础上,还要对合并的最终结果选取适当的策略,如剪枝、局部重新比对和随机选取序列重新比对进行迭代,直到比对精确收敛或者迭代次数达到上限。迭代式比对策略可以解决渐进式比对初期可能遗留下的问题。因为渐进式比对策略是贪心策略,在初期局部的比对结果上可能陷入局部最优,而错误会一直保留至最终结果中。而通过迭代式比对可以选取适当的策略,去更正局部比对的一些错误,但迭代式比对增加了时间复杂度。这两者都有着较高的时间复杂度,所以难以在有限时间内处理大规模数据的比对。
渐进式比对策略的时间复杂度与序列数量呈多项式级增长,因此在面对大规模数据的情况下,该策略时间复杂度太高、比对时间过长。而星比对是一种启发性的策略,其时间复杂度与序列数量呈线性增长,这有效降低了大规模序列比对的时间。然而,星比对算法在相似度不高的数据集上的比对精度较低,目前只能应用到相似度非常高的同源序列上,这大大限制了星比对的应用。
针对星比对精度低的问题,本文将渐进比对的模式应用于星比对中,提出了基于profile比对的改进星比对算法。实验证明改进后的算法提高了比对的精度,同时也节省了比对时间。
-
本研究改进的星比对算法采用了渐进比对的思想,先构建一颗比对的指导树,然后沿着树的枝干进行比对。但是与传统的构建指导树的方法不同,本研究沿用了星比对的核心思想,即中心比对的思想。为了加快比对速度,引进了k-band策略以加速双序列之间的比对。
-
对于双序列比对问题,主要采用动态规划的方法,有全局比对Needleman-Wunsch算法[9]和局部比对Smith-Waterman算法[10]。
图1展示的是全局比对算法,即先建立一个得分矩阵然后根据计算规则计算最大得分(匹配+1、不匹配−1、空格−1),再从右下角的最大得分回溯至左上角,得到最优比对。

图 1 DNA双序列比对动态规划算法
本研究采用的是仿射罚分与k-band结合的双序列全局比对算法[8]。不同于图1的线性罚分,仿射罚分的机制更为合理,这有效地避免了间断出现缺失的情况,使得比对结果更倾向于连续出现缺失,这也更符合生物学的进化过程,即一旦缺失位点出现,那么此位点就会有更大可能性再次出现缺失。k-band策略指的是两条序列较为相似时,回溯路线一般会在对角线附近,非对角线附近区域的值可以不用计算,只用计算对角线附近的宽为
$ k $ 的带,这个宽为k的带被称为k-band。k-band这一启发性策略减少了比对的时间和空间复杂度,将时空复杂度由$ O\left(pn\right) $ 降低到了$ O\left(kn\right) $ ($ p\mathrm{和}n $ 为序列的长度,$ k $ 为带的宽度)。如图1所示,回溯路径只出现在$ k=1 $ 的灰色带中。$ k $ 的初始值一般为$ p $ 和$ n $ 的差值的绝对值,然后进行k值的迭代,计算比对的最优得分,每次迭代$ k $ 值翻倍,直到得分最大值收敛则停止迭代。虽然采用k-band策略的双序列比对算法可以减少算法的时间和空间复杂度,但是不能确保找到最优的比对,有可能会陷入局部最优解中。为了减少此类情况的发生,只对序列长度相近的序列采取k-band策略的比对,对于序列长度相差较大的序列则采取全局比对的方法。因为两条序列长度相差较大,可能会导致k值迭代后的k-band的区域很大,甚至超过原本
$ {p}{n} $ 大小的区域面积,不仅无法节省时间和空间,反而需要更大的时空复杂度。 -
传统的星比对算法主要分为以下3个步骤:
1)选取中心序列;
2)将中心序列与其余序列一一进行比对;
3)根据“once gap, always gap”的原则,将双序列比对的结果合并,得到最终的比对结果。
中心序列的选取是传统星比对算法步骤中时间复杂度最高的,因为需要计算两两序列之间的相似度。传统计算序列两两相似度的方法是动态规划,时间复杂度为
$ O({n}^{2}) $ 。但是由于其复杂度太高,目前使用较为广泛是使用k-mer法计算序列相似度,其时间复杂度为$ O\left(n\right) $ 。使用k-mer计算序列间的相似度,选取中心序列的总体复杂度可降为$ O({m}^{2}n) $ ,其中m为序列的条数,n为序列的长度。步骤2)需要将中心序列与其余序列一一做比对,其算法时间复杂度为$ O\left(kmn\right) $ 。步骤3)合并序列比对结果的算法时间复杂度为$ O\left(mn\right) $ 。结合3个步骤的算法时间复杂度,该算法的总体时间复杂度为$ O({m}^{2}n+kmn) $ 。 -
本研究对传统的星比对算法进行了改进。首先,参考了cd-hit[11]聚类软件的思路,将最长的序列作为中心序列。然后,引进了渐进比对的思想,将构建指导树和profile比对的策略加入到改进的星比对中来。
改进的星比对算法主要有以下4个步骤:
1)选取最长序列;
2)计算最长序列与其余序列之间的相似度;
3)根据步骤2)得到的相似度构建比对的指导树。构建指导树的原则为,先将相似度最高的序列聚合,再依次根据相似度,将序列加入树中,最终构建一颗单链指导树;
4)沿着指导树进行比对和合并,最终得到比对结果。
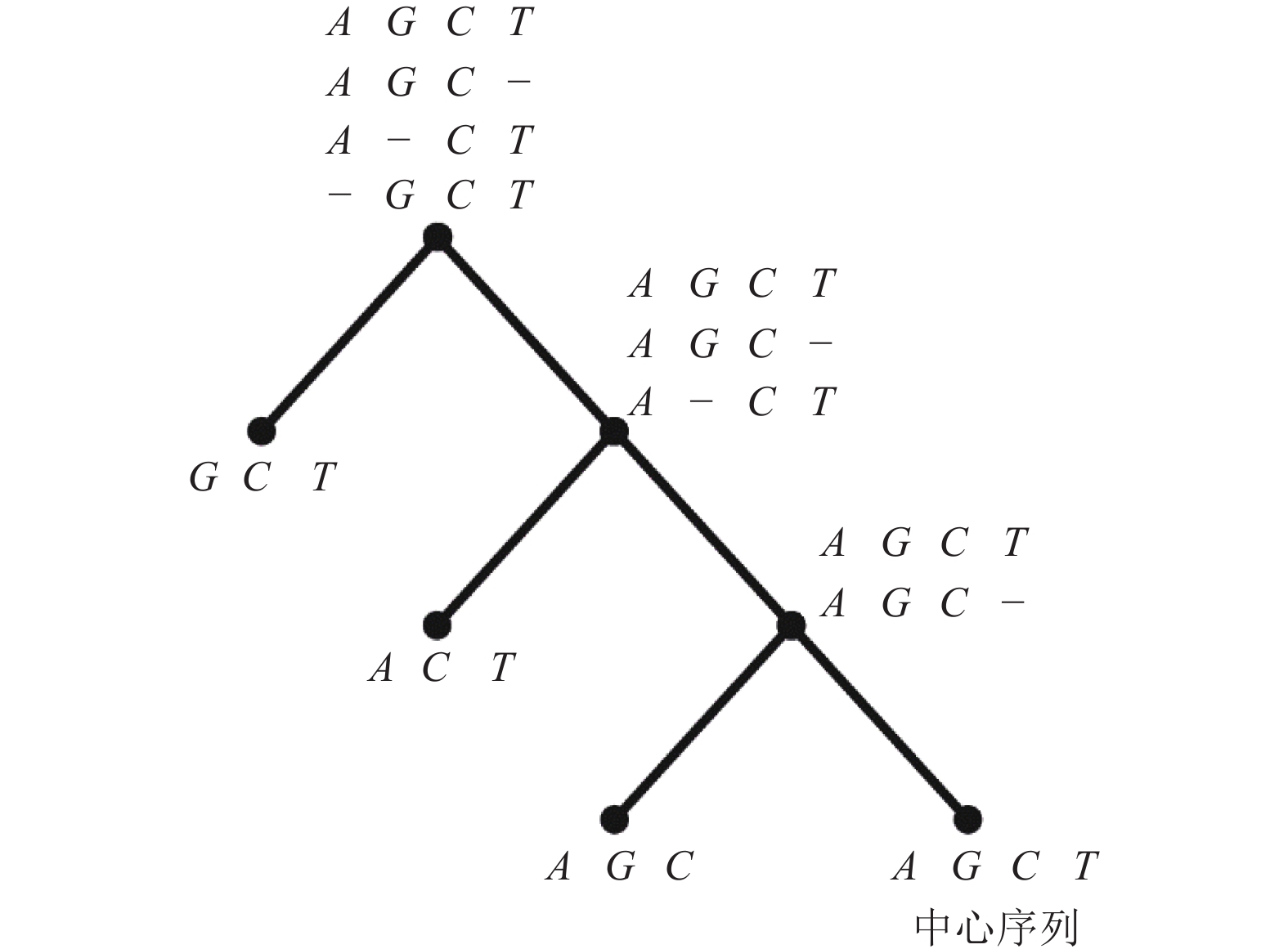
图2展示了构建指导树和比对的过程。本研究将序列中的最长序列作为中心序列,这大大降低了选取中心序列的时间。改进后的星比对算法,双序列比对只会在首次比对的时候出现,在指导树的枝干上都是序列与profile比对。与双序列比对不同,序列与profile比对计算得分耗时会更长,在一定程度上会增加比对的时间。因此,改进后的星比对算法的时间复杂度与序列数量不呈现线性增长,其增长速度介于渐进比对和传统星比对之间。

图 2 4条DNA序列构建指导树
改进的星比对算法选取“中心序列”只需要
$ O\left(m\right) $ 的时间复杂度。步骤2)需要计算最长序列与其余序列的相似度,本研究采用k-mer方法,步骤2)的时间复杂度为$ O\left(mn\right) $ 。步骤3)构建指导树,需要先将序列的相似度排序,因此步骤3)的时间复杂度为$ O\left(m{\rm{log}}m\right) $ 。步骤4)是沿着指导树进行序列和profile的比对,其时间复杂度为$ O(kmn+{m}^{2}n) $ 。结合4个步骤的算法时间复杂度,即改进的星比对算法时间复杂度为$ O(kmn+{m}^{2}n) $ 。 -
本研究采用了模拟的RNA数据来验证算法的有效性。根据序列数量将数据集分5个组,序列数量分别为:256条、512条、1024条、2048条、4096条,序列平均长度约为1500个碱基对。每个组分别有20个不同数据集,测试多数据集以求取精度的平均值,可以验证算法的鲁棒性,避免因为偶然性影响实验结果的有效性。实验数据来自于公开数据集网站
https://kim.bio.upenn.edu/software/csd.shtml .实验采用了SP score来衡量多序列比对的效果,该值是多序列比对中所有双序列组合的比对得分之和。双序列计算得分规则为:相同位置字符匹配则得1分,不匹配或者两者都是空格则得0分。SP分值越高则代表比对的效果越好。而在数据较大的时候,SP值过大不能准确展示其精度,因此本研究采用了SP值的平均值来展示比对精度。
本研究对传统的星比对算法做了两项改进:1)改进选取“中心序列”的策略,以降低选取算法的时间复杂度;2)引进了渐进比对的思想,构造一棵特殊的指导树,加入profile比对的策略。为了研究两项改进对比对精度的影响,本文设计了4种实验的算法组合:传统的星比对算法+传统的中心序列选取策略、传统的星比对算法+选取最长序列、改进的星比对算法+传统的中心序列选取策略、改进的星比对算法+选取最长序列,比对4组实验的精度和时间。4组实验都是在CPU为Xeon E3-1230,内存为32 G的Ubuntu 20.04系统环境下进行的。
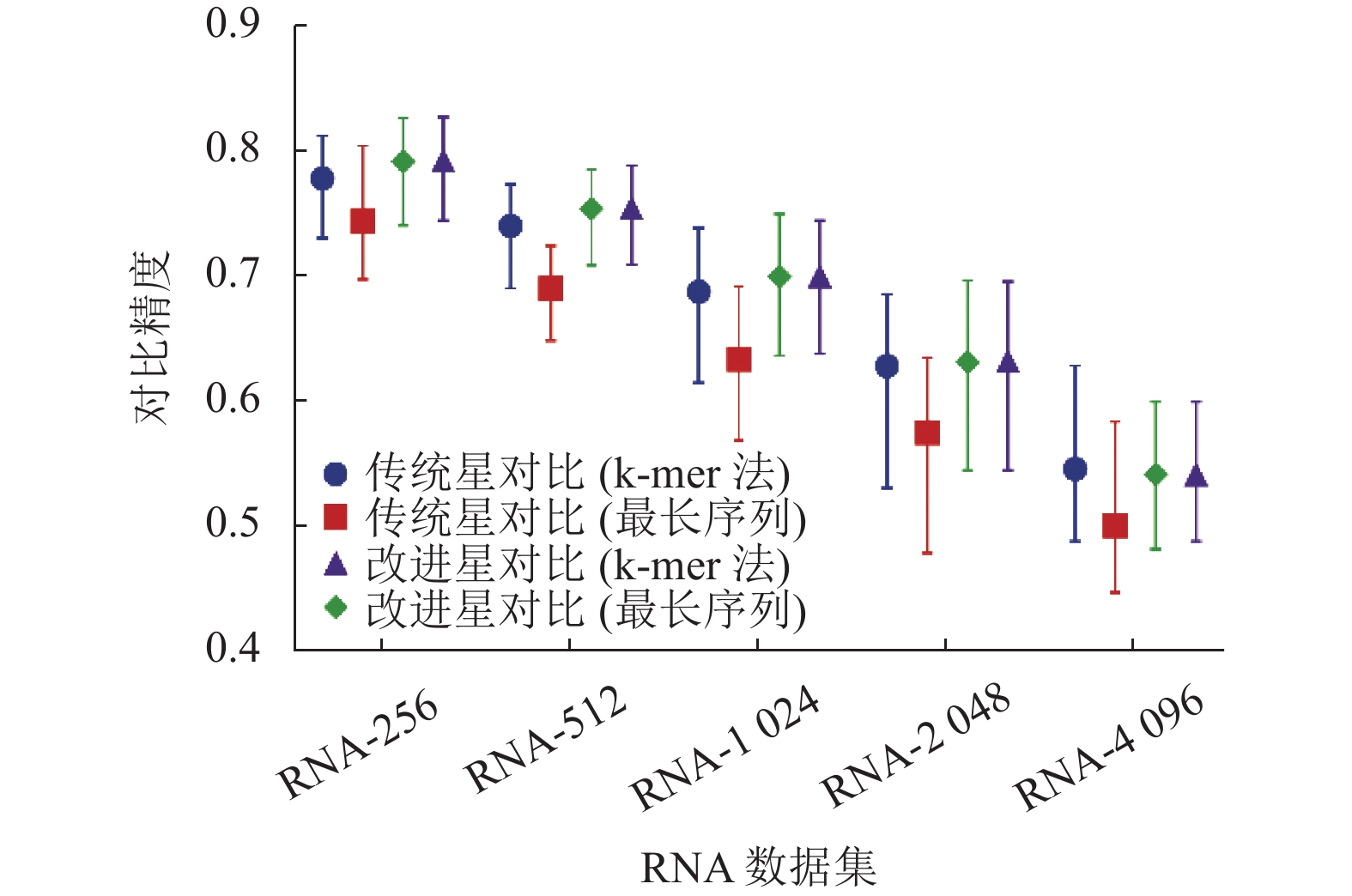
实验结果如图3和图4所示。图3展示了4组算法在不同数据集上的精度。可以看到随着数据集的增大,比对的精度也随之降低。

图 3 不同数据集的比对精度对比
在使用传统的星比对算法的基础上,对比使用不同的选取中心序列的策略。使用k-mer法计算相似度选取中心序列的策略明显优于使用最长序列计算相似度的策略,可以观察到在5组数据集中,使用最长序列策略的比对精度都要比传统的策略低。而在使用改进的星比对算法的基础上,两种选取中心序列的策略得到的比对效果近乎一致,无论是在比对精度的均值还是在比对精度的范围上,两者并无明显差异。
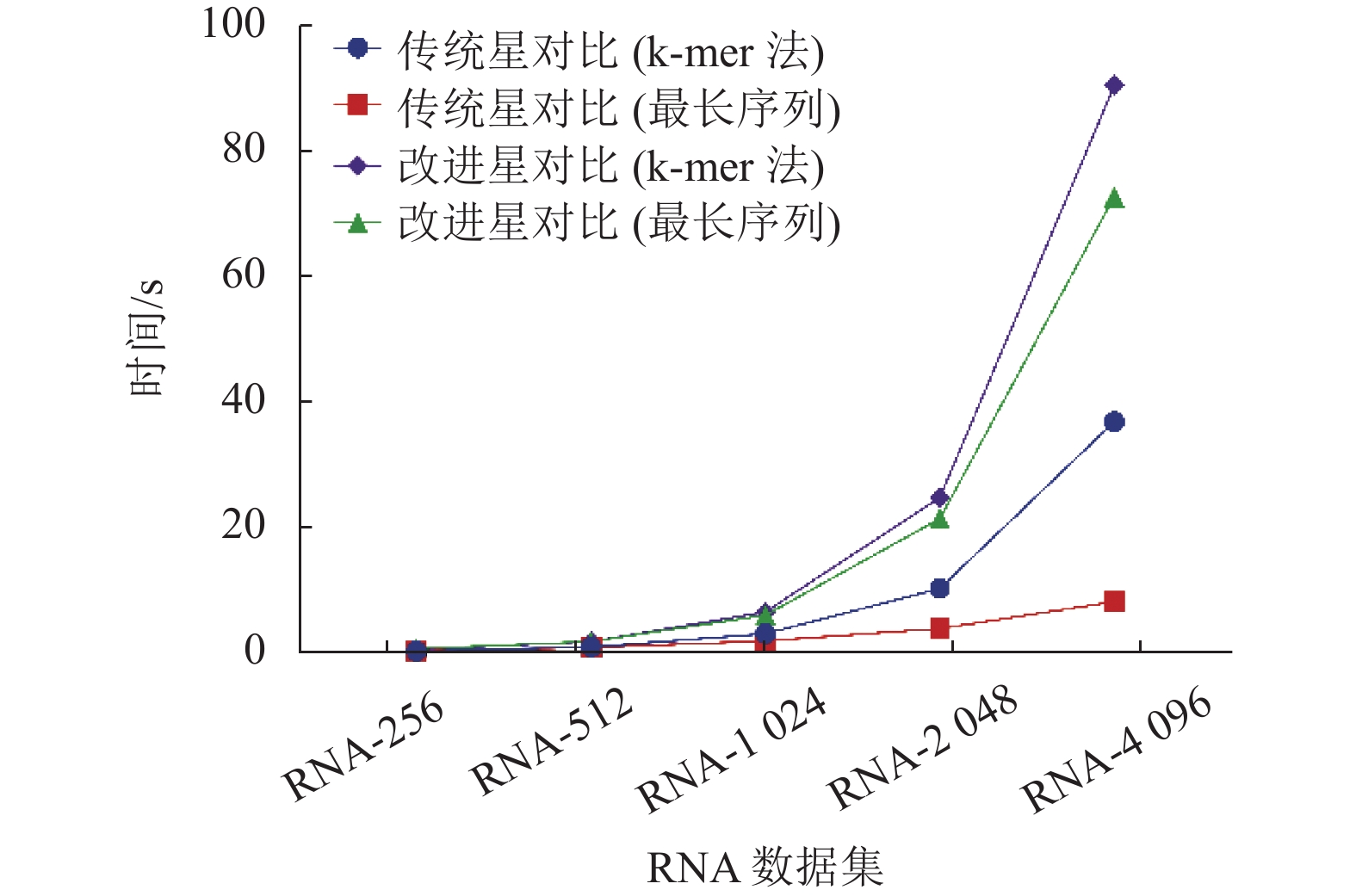
图 4 不同数据集的比对时间对比
在使用传统策略选取中心序列的基础上,改进的星比对算法的比对精度明显要优于传统的星比对算法。同样,在使用最长序列作为中心序列的基础上,改进的星比对算法的比对精度也明显优于传统的星比对算法。
由图3可知,在4组实验中,可以看到改进的星比对算法的比对效果优于传统的星比对算法。从图4可知,使用最长序列作为中心序列的策略,可在一定程度上减少比对的时间。因此改进的星比对算法加上使用最长序列作为中心序列的策略是最佳的组合方式,此组合可以得到最好的比对精度,同时不会显著提升星比对的比对时间。
-
本文将传统的星比对与渐进比对相结合,提出了基于profile比对的改进星比对算法,改进后的星比对算法显著提高了比对的精度。为了减少比对时间,本研究还简化了中心序列的选取,直接将最长序列作为中心序列。改进前后的算法时间复杂度是一致的,但实际时间不一定一致,改进的星比对算法运行时间要略大于传统的星比对算法。同时,两者运行时间随着数据量级增大的增长速度是一致的。由此可见,本文提出的基于profile比对的改进星比对算法不仅提高了比对的精度,又通过简化中心序列的选取减少了星比对中选取中心序列的时间,同时也并未增加比对算法的时间复杂度。
Improved Center Star Alignment Algorithm Based on Profile Alignment
-
摘要: 多序列比对在序列分析研究中起着重要的作用,包括功能重要位点的识别和系统发育分析等问题。目前大多数比对软件都使用渐进比对或迭代比对的策略,但两种策略都具有较高的时间复杂度,因此难以处理长序列和大规模序列的比对问题。而星比对虽然具有很低的时间复杂度,但精度并不理想,目前只适用于相似度非常高的序列。针对此问题,引进了渐进比对中的profile比对来改进星比对算法的精度,同时避免大幅度地增加星比对的时间复杂度。最后,通过实验证明了改进的星比对算法可以有效地提高比对的精度。Abstract: Multiple sequence alignment plays an important role in sequence analysis, including identification of functionally important sites and phylogenetic analysis. At present, most alignment software uses the strategy of progressive alignment or iterative alignment, but both strategies have high time complexity, so it is difficult to deal with the alignment problem of long sequence and large datasets. Although star alignment has a very low time complexity, the accuracy of star alignment is not ideal, so it only applies to sequences with very high similarity. To solve this problem, we introduce profile alignment in progressive alignment to improve the accuracy of star alignment algorithm and avoid significantly increasing the time complexity of star alignment. Experiments show that the improved star alignment algorithm can effectively improve the accuracy of the alignment.
-
[1] BISWANATH C, GAUTAM G. A review on multiple sequence alignment from the perspective of genetic algorithm[J]. Genomics, 2017, 109(5): 419-431. [2] KATOH K, MISAWA K, KUMA K, et al. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform[J]. Nucleic Acids Research, 2002, 30(14): 3059-3066. doi: 10.1093/nar/gkf436 [3] LASSMANN T. Kalign 3: Multiple sequence alignment of large datasets[J]. Bioinformatics, 2020, 36(6): 1928-1929. [4] SIEVERS F, WILM A, DINEEN D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega[J]. Molecular Systems Biology, 2011, 7(1): 539. doi: 10.1038/msb.2011.75 [5] THOMPSON J D, HIGGINS D G, GIBSON T J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice[J]. Nucleic Acids Research, 1994, 22(22): 4673-4680. doi: 10.1093/nar/22.22.4673 [6] EDGAR R C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity[J]. BMC Bioinformatics, 2004, 5(1): 1-19. doi: 10.1186/1471-2105-5-1 [7] NOTREDAME C, HIGGINS D G, HERINGA J. T-Coffee: A novel method for fast and accurate multiple sequence alignment[J]. Journal of Molecular Biology, 2000, 302(1): 205-217. doi: 10.1006/jmbi.2000.4042 [8] ZOU Q, HU Q H, GUO M Z, et al. HAlign: Fast multiple similar DNA/RNA sequence alignment based on the centre star strategy[J]. Bioinformatics, 2015, 31(15): 2475-2481. doi: 10.1093/bioinformatics/btv177 [9] NEEDLEMAN S B, WUNSCH C D. A general method applicable to the search for similarities in the amino acid sequence of two proteins[J]. Journal of Molecular Biology, 1970, 48(3): 443-453. doi: 10.1016/0022-2836(70)90057-4 [10] SMITH T F, WATERMAN M S. Identification of common molecular subsequences[J]. Journal of Molecular Biology, 1981, 147(1): 195-197. doi: 10.1016/0022-2836(81)90087-5 [11] FU L M, NIU B F, ZHU Z W, et al. CD-HIT: Accelerated for clustering the next-generation sequencing data[J]. Bioinformatics, 2012, 28(23): 3150-3152. doi: 10.1093/bioinformatics/bts565 -
 点击查看大图
点击查看大图

图(4)
计量
- 文章访问数: 4308
- HTML全文浏览量: 1481
- PDF下载量: 57
- 被引次数: 0






















