 ISSN
ISSN 


-
2005年《Science》杂志在创刊125周年之际,发表“能否预测蛋白质折叠?”,被列为21世纪125个最具挑战性的科学前沿问题之一[1]。蛋白质分子机器如何自发地组装形成特定功能结构是生物学中心法则完整图景中一个最为关键的遗留问题,是生命科学领域尚未解决的重大基础性科学问题之一。
1994年,美国马里兰大学的Moult课题组创立了世界性的蛋白质结构预测竞赛CASP(critical assessment of structure prediction),进行两年一度的盲评估,以促进研究、监控进展,并确立蛋白质结构预测的最新水平[2]。CASP测试蛋白分为基于模板(template based modeling, TBM)和无模板(free modeling, FM)两类。TBM类可以将PDB(protein data bank)结构数据库中的已有实验结构作为模板进行同源建模,其建模精度与实验测定水平相仿[2]。相对而言,缺乏同源模板的FM类蛋白难度更大,更具有挑战。受限于能量模型不精确性和构象空间采样瓶颈[3],从CASP5(2002年)到CASP10(2012年)10年间,FM预测方法陷入了长期的发展停滞期[4]。2014年,共进化方法被引入CASP11接触预测组,接触预测准确性出现了进步的迹象[5]。至此,结合共进化接触预测的构象采样方法成为FM预测的主流。经过两年的发展,尤其是深度卷积残差网络ResNet的首次应用[6],在2016年的CASP12中,接触预测精度提升至47%[7]。2018年的CASP13中,通过将接触拓展为残基间距离,接触残基对的预测精度达到70%[7-8]。在蛋白质接触图及距离深度学习预测技术进步的推动下[5],在CASP13中,FM类目标蛋白的平均GDT_TS(global distance test total score)超过了60。
在2018年的CASP13中,Google的DeepMind团队凭借其开发的AlphaFold在43个FM类目标蛋白中拿到25个单项最佳模型,并获得总分第一名[8]。在2020年的CASP14中,Google的DeepMind团队开发的第二代人工智能(artificial intelligence, AI)蛋白质结构预测程序AlphaFold2[9],在中等难度目标蛋白上基本达到实测测定结构精度。CASP评委Andrei Lupas教授在接受《自然》杂志的采访中讲道[10]:“它将改变医学、改变研究、改变生物工程、改变所有!”。随后,华盛顿大学Baker课题组开发了一种新的AI蛋白质结构预测三轨网络RoseTTAFold[11],预测精度接近AlphaFold2。由于AlphaFold2和RoseTTAFold在蛋白质结构预测领域的突破,蛋白质结构预测算法被《Nature Methods》杂志评选为“2021年度方法”。AI预测蛋白质结构显然是结构生物信息学领域的重大突破,但是正如著名生物学家、斯坦福大学Brunger教授在《Science》杂志上发表的论文中指出,蛋白质结构预测问题距离“解决”仍然很远[12]。
通过对蛋白质组学数据的分析,2003年剑桥大学Chothia等人在《Science》杂志发文指出:自然界生物中大约有超过80%的真核蛋白和67%的原核蛋白含有多个结构域[13]。2019年8月本课题组对PDB库的统计结果也表明[14],在17多万个实验结构测定蛋白中共包含了608044个单链结构,其中只有34.7%的单链为多域蛋白。考虑到PDB库中存储的蛋白结构均为实验测定这一事实,可以得到一个明显的结论:由于X衍射、NMR(nuclear magnetic resonance)及冷冻电镜等结构生物学实验测定手段的技术瓶颈,多域蛋白结构实验测定速度远远低于单域蛋白。而AlphaFold2和RoseTTAFold预测过程中不但使用了同源序列信息,还包括了结构模板信息。这意味着对于多结构域全长链蛋白,当无法检测到全长链模板时,或者每个结构域具有不同的同源序列时,直接通过单结构域的方式预测全长链多结构域蛋白效果并不理想[7]。
2019年Moult发表的CASP13综述论文指出,具有多个单结构域的全长链建模(即多域蛋白质结构预测)将会是未来CASP竞赛的发展趋势[2]。本文结合国内外研究现状以及本课题组开展的一些研究工作,针对多结构域蛋白质结构预测方法的研究进展进行分析和综述。
-
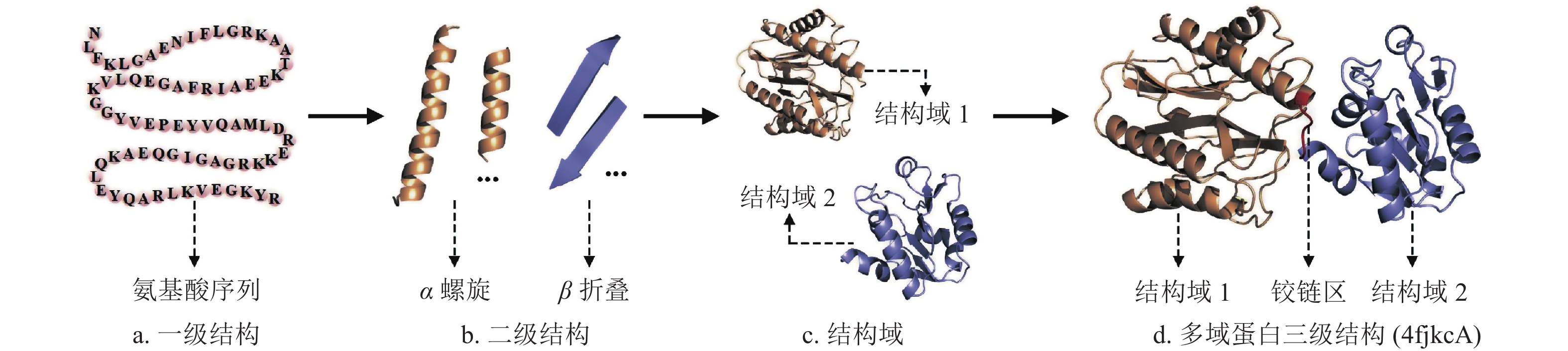
结构域(domain)是位于二级结构和三级结构之间的一个层次,一般由100~250个氨基酸残基组成。结构生物学领域普遍认为结构域是蛋白质三级结构内的独立折叠单元[13]。一条较长的蛋白质全长单链通常会包括若干个结构域,某些区域相邻的氨基酸残基首先形成有规则的二级结构,然后由若干二级结构折叠成近似于球状的结构域,最后通过两个或多个结构域组装形成多结构域蛋白(多域蛋白)的三级结构。图1给出了多域蛋白(PDBID:4fkcA)的一级序列结构、二级结构、结构域、多域蛋白空间折叠过程的示意图。
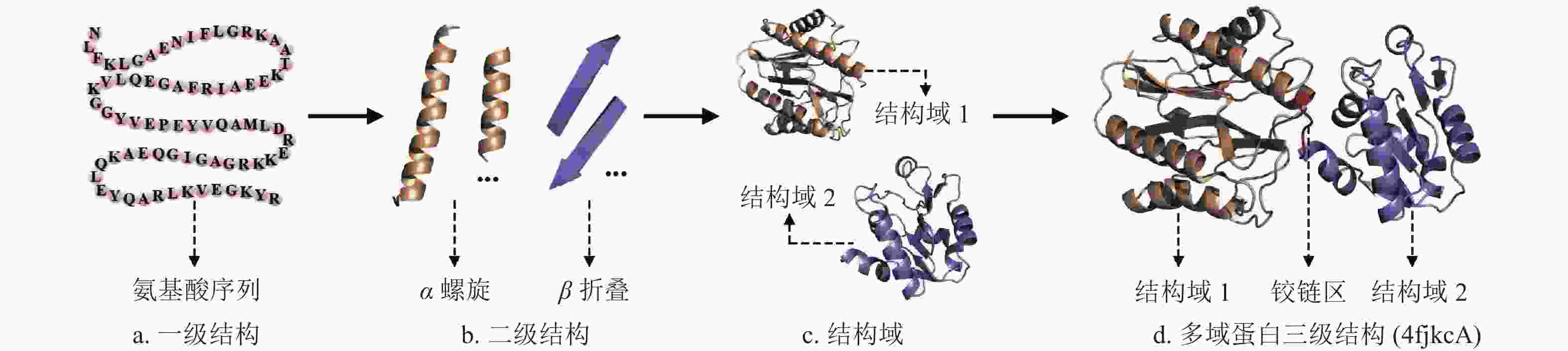
图 1 多域蛋白空间折叠过程示意图(PDBID: 4fkcA)
同时,结构生物学领域也普遍认为结构域是一个独立的功能单元,承担着独立的生物学功能。然而,从生物学意义上来讲,结构域间缔结对于促进以协作方式实现多个相关的功能至关重要,如糜蛋白酶功能就是通过两个结构域之间接触面所形成的活性位点来完成。在多域蛋白中,连接两个结构域之间铰链区(linker)的结构柔韧性,使结构域间容易发生相对运动,这将有利于结合底物或施加应力,有利于别构中心结合调节物并发生别构效应,以利于酶对反应的催化,这些部位往往是活性中心所在的部位,或是变构物结合的部位。因此,阐明多域蛋白的结构有助于理解其所具备的重要生物学功能。
-
蛋白质结构预测一直受到计算生物学领域和计算智能社区的高度关注,是一个前沿研究课题。其中具有代表性的有:张阳课题组开发的I-TASSER系列[15]连续八届(CASP7~CASP14)在CAS服务器组排名第一;谷歌DeepMind开发的AlphaFold系列自从2018年首次进入结构预测领域后,连续两届在CASP人工组排名第一[8];Baker课题组尽管在过去几年中工作重点已投入到从头设计蛋白质,然而,开发的Rosetta系列[16-17]二十多年来在结构预测领域得到了“教科书”式的广泛应用,2020年CASP14中人工组排名仅次于AlphaFold2;许锦波课题组第一次将ResNet应用在蛋白质接触预测[6],真正推动了深度学习在蛋白质残基接触、残基间距离的应用,是蛋白质预测领域发展的里程碑,在2018年的CASP13中,许锦波团队开发的RaptorX系列在Contact组名列前茅[7]。
总之,AlphaFold系列、I-TASSER系列、Rosetta系列、RaptorX系列等方法和服务器基本代表了蛋白质结构预测领域近五年来最先进的主流预测技术。从中也不难发现,无模板方法与基于模板方法、传统理化能量模型与深度学习知识能量模型、片段采样和几何优化方法之间的界限越来越模糊,它们之间相互融合,共同促进。这使得对所有这些方法进行严格分类并不是一件容易的事情。本文主要针对单域蛋白结构预测方法、多域蛋白结构组装方法以及端到端的单体蛋白预测方法3部分展开讨论。单域蛋白结构预测方法主要按照基于理化知识模型的模拟方法和基于深度学习模型的能量极小化方法两类进行分析。
-
这类方法的基本思想是首先建立蛋白质主链粗粒度和全原子细粒度表达模型,综合考虑分子间物理化学作用及从蛋白质序列库、结构库推断出的结构特征知识,分别构建基于理化知识的粗粒度和细粒度蛋白质能量数学模型。其次,基于粗粒度能量模型,设计构象空间优化方法搜索能量函数的全局最优构象,进而在细粒度模型能量函数的引导下,对全局最优构象进行结构精修。代表性的能量模型主要有I-TASSER、QUARK[18]及Rosetta[16-17]。模型优化方法主要包括:Metropolis蒙特卡罗极小化 (Metropolis Monte Carlo, MMC)[19]、副本交换蒙特卡罗 (replica exchange Monte Carlo, REMC)[20]、分子动力学模拟(molecular dynamics, MD)[21]及进化算法[3, 22-23]等。
此外,针对蛋白质构象空间的高维特性,片段组装策略利用已测定蛋白质结构的局部信息,将连续的二面角度构象空间优化问题转变成了离散的片段组合优化问题,有效地减少了构象搜索空间,对基于理化知识模型的模拟方法发展起到了巨大的推动作用[15-16, 24]。从1994年CASP1到2020年CASP14的26年间,基于理化知识模型的模拟方法在CASP竞赛中占据了支配性地位。在2020年底结束的CASP14竞赛中,I-TASSER在服务器组中排名第一,Rosetta在人工组中排名第二(第一名为AlphaFold2[9])。
I-TASSER[25-26]和QUARK[27]是两款蛋白质结构预测服务器。I-TASSER使用穿线方法识别PDB库中的结构模板,基于结构模板构建蛋白模型;QUARK则是在理化知识能量模型的引导下,采用REMC[20]方法对序列上长度为1~20个残基的特定位置进行片段组装生成蛋白模型。张阳课题组还整合了QUARK与I-TASSER两种方法,论证了模板建模和无模板建模方法的结合可以有效提升从头预测的精度[28]。在2016年的CASP12中,通过在QUARK中加入NeBcon[29]预测的接触约束,前5个预测模型平均TM-score为0.41,相对于不使用接触约束的QUAKR模型高出了37%[30],这表明接触约束可以有效提升FM目标蛋白的预测精度。在2018年的CASP13中,采用新开发的序列比对生成协议,进一步将基于深度学习的接触预测方法ResPRE[31]集成到NeBcon元方法中,有效提升了蛋白残基间接触预测的精确度。同时,对指导结构采样的接触势能做了进一步优化,推出了C-I-TASSER[32]和C-QUARK[33]两个版本。对于CASP13中的50个FM目标域蛋白,C-I-TASSER和C-QUARK构建的第一个模型平均TM-score分别比I-TASSER和QUARK的构建模型高出28%和56%。而且,第一次证明了接触预测在TBM目标域蛋白上的有效性[15]。在2020年的CASP14中,D-I-TASSER和D-QUARK再次在服务器组中拔得头筹。性能提升的关键在于3个方面:引入深度学习算法精确预测氨基酸间距离和氢键;利用I-TASSER平台将穿线模板与深度学习预测的距离和氢键约束有机结合;使用宏基因组构建高质量的多序列比对。
Rosetta[16-17]是生物大分子建模软件,集成了蛋白质结构建模和分析的各种采样算法和能量函数。除了蛋白质结构预测Rosetta Abinitio模块之外,还提供了从头蛋白质设计、酶设计及分子对接等功能。Rosetta提供一个灵活的功能库来完成各种生物分子建模任务。这些库定义的基本任务和操作作为算法被组合在一起,称为“Protocols”,每种Protocol都使用Rosetta的分子建模库来完成特定的建模任务。这些协议可以用作独立单元,也可以将它们链接在一起以完成更复杂的任务,如可以在通用框架内组合Protocols。这些特征使得Rosetta在蛋白质结构预测领域得到了广泛应用,极大地推动了蛋白质结构预测领域的发展。CASP11竞赛中,从共进化分析得到的残基间接触被用作作为约束条件,使得预测的模型质量普遍得到提高。之后,Baker意识到如果一个蛋白质家族有足够多的序列,则可能根据进化期间的共变现象来推断出残基间的接触关系。但是,如果目标蛋白没有足够的多样性多序列比对(multiple sequence alignment, MSA)时,是否可以通过宏基因组获取呢?基于上述思想,2017年Baker课题组首次将宏基因组数据整合在Rosetta中,相关工作发表在《Science》杂志上[34]。该研究结果表明,基于宏基因组数据可以产生精度更高的接触信息,结合基于接触的结构匹配和Rosetta采样方法,成功预测了614个未知结构的蛋白质模型。这一工作对蛋白质结构预测领域有着深远影响,在2020年的CASP14上,可以看到AlphaFold2、I-TASSER、QUARK等最先进的预测方法中均利用了宏基因组的数据信息。同年,文献[35]研究发现在超过4000个蛋白质家族中,有25%的直接共进化残基对在三维结构上距离超过5Å,3%残基对三维空间结构距离超过15Å。这一发现为2018年开始兴起的距离预测提供了重要的理论依据。
-
这类方法的基本思想是针对特定查询蛋白序列,首先,通过序列比对方法对蛋白序列(或宏基因组)数据库进行搜索生成多序列比对(MSA)序列集合。然后,基于共进化原理,通过深度学习方法分析MSA中的协同进化模式,推断出三维结构空间中残基间的接触、距离、方位等空间约束条件。最后,基于空间约束条件直接构建数学模型,通过优化方法直接求解得到蛋白质三维结构模型。2011年,文献[36]在基于共进化方法预测蛋白质结构的挑战方面迈出了一大步,并在随后的CASP11~CASP14中得到广泛验证。2012年,程建林课题组最先把深度学习技术应用到共进化分析方面[37],彻底改变了传统的接触、距离预测技术[38-39]。2017年,许锦波课题组首次将深度卷积残差神经网络(ResNet)应用到共进化分析接触预测,第一次真正意义上展现了深度学习在蛋白质结构预测领域的巨大力量[6]。这些事件都是蛋白质结构预测领域的里程碑。
深度学习方法经过了2012年CASP10的萌芽阶段,2014年CASP11的验证阶段,2016年CASP12的发展阶段和2018年CASP13完善阶段之后,在2020年的CASP14中最终取得了重大进展和突破。DeepMind开发的AlphaFold2[9]通过端到端的深度学习,甚至可以直接从序列学习到蛋白质精确的三维结构。CASP竞赛发起者之一,Moult在2019年指出[2]:“在蛋白家族中有足够数量序列的前提下,最新(指深度学习)预测方法基本上解决了长期以来单结构域蛋白质折叠拓扑结构预测的难题,而且比对所需要的序列数量已大幅下降,同时对基于模板建模方法的准确性也有了实质性的提高。”深度学习方法似乎可以有效地集成关于共同进化残基对、片段之间相互作用信息,或者利用序列相似性的记忆信息,有时甚至可以在几乎没有任何目标特定进化信息的情况下提供准确预测结果[5]。
许锦波课题组在ResNet接触预测的思路上往前推进了一步。他们发现距离(连续值)比接触(0或1)更有用,通过深度学习整合模板和共进化信息,可以有效改善蛋白质结构建模质量。在CASP13中,许锦波课题组开发了基于距离的接触预测、穿线及折叠方法的RaptorX 3个服务器版本。在32个CASP13 FM目标上,RaptorX在46个参赛组中获得最佳接触预测排名,也是服务器组中最好的三维结构预测服务器之一。RaptorX在前L/5、L/2和L远程接触预测精度分别达到了70%、58%和45%,在所有参赛组中得到了T0950-D1和T0969-D1的最佳三维结构预测模型[7]。同一时期,2018年DeepMind首次参加CASP13三维结构预测人工组竞赛,并推出了蛋白质结构预测一代产品AlphaFold[8]。AlphaFold采用了与学术界近乎同样的方法(或是同期并行开展),通过训练ResNet网络学习距离约束,进而构建距离约束数学模型,通过拟牛顿优化方法求解结构模型,并且不对多域蛋白进行分割,直接进行全长链建模。AlphaFold在人工组中累计总分68.3,排名第一(排名第二为张阳实验室I-TASSER系列,总分为48.2),并在43个目标蛋白质中获得了25个单项最佳模型。AlphaFold的实质性进展成功地表明通过简单的几何优化方法,辅以高精度的距离预测约束,是一种行之有效的蛋白质结构预测方式。在之后的一年中,文献[16]进一步将接触、距离预测扩展到方位预测,并将其集成到Rosetta能量模型中,并采用能量几何极小化方法求解结构模型,开发出了trRosetta。预测的方位相对于距离/接触而言,包含了不对称信息,能够有效避免数据的不一致性问题,在精度和效率方面基本与AlphaFold持平。随后,杨建益课题组进一步改进网络架构并加入模板开发了trRosettaX[40],在CASP14的盲测中被评为顶级服务器组之一。
同时,几个研究团队在CASP13上提出并开发了相应基于深度学习的接触预测和三维结构预测方法与服务器。文献[41]分别基于协方差、精度和伪极大似然估计建立3个谱矩阵,作为深度残差卷积神经网络结构的输入特征,用于接触图训练和预测。通过端到端的训练和叠加,提出了两种集成矩阵特征的整合策略,开发了两个互为补充的接触图预测服务器TripletRes和ResTriplet。文献[39]开发了蛋白质结构预测系统MULTICOM的增强版本。MULTICOM增强版本主要包括基于深度卷积神经网络的残基−残基对距离预测、距离驱动的自由模板建模以及基于深度学习和接触预测技术的蛋白质模型质量评估等3个部分。文献[42]基于序列比对的改进方法和扩展数据源,设计开发了一种基于深度学习的接触预测工具DeepMetaPSICOV (DMP)。在原先MetaPSICOV和DeepCov算法的基础上,DMP融合两种算法的输入特征,并将之作为深度全卷积残差神经网络的输入特征。此外,2020年,文献[43]提出了一种接触预测方法AmoebaContact,并设计了基于梯度下降的GDFold方法求解接触约束模型,通过修改AmoebaNet的NAS(neural architecture search)算法,自动搜索神经网络架构来完成接触图预测任务。
-
整体来讲,目前在蛋白质结构预测领域,包括14届CASP竞赛在内,主要还是关注于单域蛋白预测问题。相对于单域蛋白而言,目前多域蛋白结构预测问题研究工作要少得多。现有文献中多域蛋白质预测方法主要分为基于铰链区采样和基于分子刚体对接两类方法。在铰链区采样方法中考虑到多域蛋白质全长肽链连接性的因素,多域蛋白预测问题可以看作是单域蛋白结构折叠过程的一个特例,即保持每个单域结构刚性,通过调整铰链区构象来实现多域组装建模。因此,用于单域蛋白质预测的能量函数和构象空间采样方法(如Rosetta)经过一定的修正可应用于该问题[11]。在分子刚体对接方法中,考虑到结构域间的相互作用和不同蛋白质链之间的相互作用非常相似(尽管在作用机理上完全不同),多域建模可以视为若干刚体结构分子(如蛋白质−蛋白质)的对接过程,可以利用分子对接算法来求解[44]。
2007年,文献[11]提出一种基于铰链区采样的两阶段多域组装方法(亦称Rosetta多域蛋白组装方法)。在第一阶段,基于Rosetta粗粒度能量模型(即侧链用质心伪原子代表),采用MC(Monte Carlo)方法对多域蛋白铰链区骨架二面角空间进行片段重组采样,并生成5000个诱饵构象。在第二阶段,首先对第一阶段生成的每个诱饵构象,结合Dunbrack Rotamer侧链库,采用MC协议重建域间接触界面氨基酸的侧链构象;然后基于Rosetta全原子能量模型,通过Rosetta标准的MC方法进行结构精修,主要包括铰链区骨架二面角微调、铰链区和接触面残基侧链组装、铰链区骨架二面角和所有残基侧链拟牛顿几何优化以及Metropolis准则生成测试构象4个步骤。76个包含两个结构域的多域测试蛋白组装结果表明,有38个多域蛋白经过两阶段组装之后得到模型RMSD<2Å,25个多域蛋白的预测精度RMSD>2Å。测试结果也表明,有13个多域蛋白质组装失败,至少有50%的多域测试蛋白并不能捕获到两个结构域正确的方位关系。结构域连续性的限制对多域蛋白质组装过程而言是一个至关重要的因素,然而该方法并没有考虑到结构域连续性的限制,也没有考虑不连续域的情况,并且超过两个结构域的多域蛋白组装在文中并没有给出相关报道。
2015年,文献[44]提出了基于铰链区采样的多域组装方法AIDA。AIDA方法采用蛋白质三维结构简约表达模型,即每个残基包括4个主链原子和1个代表侧链中心的伪原子,其中侧链中心伪原子的位置根据骨架几何特征估计。在结构域组装过程中,每个结构域作为刚体分子,通过调整铰链区二面角改变多域蛋白的构象。在QUARK能量函数[27]基础上,进一步设计多域蛋白结构域间相互作用能量函数,考虑到单链连通性和结构域刚性约束的限制,使用了单轨迹能量极小化算法实现构象空间采样。测试集包括了136个连续2-域蛋白、36个连续3-域蛋白、13个连续3-域以上的蛋白以及20个含有不连续结构域和插入结构域的2-域蛋白。测试结果表明,独立解析结构域组装与从多结构域蛋白解析结构中提取单结构域组装相比,生成良好模型的成功率从65%下降到54%。这表明单域结构微小的变化都可能会对多域模型质量产生极大影响。此外,通过能量函数选择正确模型的成功率也从83.0%降低到53.8%,这表明设计的多域蛋白能量模型仍然还有很大的改进空间。
2019年,文献[14]提出和开发了第一个真正意义上自动化的多域蛋白质组装方法和服务器DEMO。DEMO基于分子对接原理,通过逐域结构比对[45]检测类似模板,进一步根据类似模板的距离谱特征构建域间方位。在包含2~7个连续和不连续结构域的356个多域蛋白测试集上,有86%的连续域测试蛋白和100%不连续域的测试蛋白组装形成了具有正确拓扑结构的全长链折叠模型。在CASP12和CASP13中的多域目标蛋白组装结果也表明,DEMO生成的全长链模型精度显著提升。进一步,引入质谱交联数据CL和冷冻电镜密度图Cryo-EM的稀疏约束,组装模型的平均TM-score又分别提高了6.3%和12.5%。测试结果表明,DEMO是一种高效自动的全长链建模方法,有进一步适用于全基因组级规模的多域蛋白组装的潜力。尽管给出了一些成功案例,DEMO在CASP14的盲测中效果并不尽人意。在以下几个方面需要进一步改进:1) 在DEMO模拟过程中域结构一直保持刚性,这不能合理地解释由于绑定引起的构象变化。此外,预测的结构域通常具有较低的分辨率,因此在域组装模拟中引入主链骨架灵活性可以为单域局部结构细化提供可能。2) 近年来,基于共进化的接触和距离深度学习预测方法在蛋白质三维结构预测领域已经取得了巨大进展和突破。借鉴这一成功经验,基于序列的域间接触和距离信息可以引入到DEMO中,进一步细化得到更为合理的域间方位。
-
基于深度学习的端到端方法抛开了传统的折叠模拟过程,直接从一级序列构建三级结构。这类方法采用深度学习网络模型直接从输入到输出(序列到结构)联合调整模型参数,在一定程度上避免了距离、方位等预测网络固有的不一致性。最具代表性的端到端方法包括第二代程序AlphaFold2[9]和结构预测端到端三轨网络RoseTTAFold[11]。
不同于第一代AlphaFold,AlphaFold2中使用一整套的注意力机制取代了以蛋白质信息构建不同氨基酸彼此接近程度的图表再建模的相对传统的方式。AlphaFold2的整体系统架构有两个主要的处理“轨道”,其中一个轨道的输入表示MSA的行和列,另一个轨道的输入本质上表示蛋白质模型中每个氨基酸之间的原子间距离。MSA路径允许网络跟踪氨基酸守恒和协变特征,而距离矩阵提供每对氨基酸的3D空间信息,这两个轨道之间还可以交换信息。这意味着随着距离信息的改进,可以重新解释MSA,在重新解释MSA时,也可以进一步改进距离信息。最后,来自两条轨道的信息被输入结构模块,该模块试图构建蛋白质的3D模型:即无需外部建模程序的情况下,直接输出氨基酸残基的3D坐标。最后,以旋转不变的特殊几何形式表示的结构将会基于注意力机制进行迭代改进。这种旋转不变性是基于结构生物信息学中的标准共价几何实现的,即在每个氨基酸周围定义局部坐标框架[46]。AlphaFold2展示了一种联合嵌入多序列比对(MSA)和成对特征的新体系结构、一种新的输出表示和相关损失、一种新的等变注意体系结构,并自我估计准确度[9],大大提高了结构预测的准确性。
RoseTTAFold[11]是受到DeepMind研究结果启发后开发的一个“三轨”(three-track)神经网络模型,与AlphaFold2在同一天分别发表于《Science》和《Nature》。在RoseTTAFold中,探索生成了一个可使信息沿着一维序列对齐轨道和二维距离矩阵轨道并行流动的“双轨”网络,其性能远远优于trRosetta。在此基础上,他们将双轨模型的两个层次与运行在三维骨干坐标上的第三个平行结构轨道相结合,从而使得1D氨基酸序列信息、2D距离信息和3D坐标信息之间能够来回流动,共同推理三者内部和之间的关系。通过RoseTTAFold(end-to-end)和RoseTTAFold(pyRosetta)的比较,他们认为侧链信息的加入可以进一步改善模型精度。
-
PDB数据库是目前最全的蛋白质结构数据库,主要收集通过X射线单晶衍射、核磁共振和电子衍射等实验手段确定的生物大分子(蛋白质、DNA 和 RNA)的三维结构。CATH[47]和SCOPe[48]是两个重要的蛋白质结构域分类数据库,且具有一定的相似之处。两者都是以自动程序和人工处理的混合方式识别蛋白质结构域进行分类,但使用不同的结构域定义和分类标准来定义结构域边界和对结构域进行分类。
文献[49]结合CATH和SCOPe数据库中定义的结构域信息,使用序列比对及结构域自动分割技术开发了MPDB,以期能为对多域蛋白质感兴趣的研究人员提供一个统一的信息门户。MPDB包含两个重要的模块:多域蛋白筛选模块和结构类似物检测模块。筛选模块根据用户输入的标准(包括蛋白链长度、分辨率、域数、Rfactor值和多域蛋白的序列一致性)对整个MPDB进行过滤后,向用户提供符合标准的蛋白质结构及相关信息。结构类似物检测模块通过单个结构域模型和MPDB库中的模板逐一进行结构比对,并根据局部−全局相似性关系识别出全链结构类似物。
-
蛋白质模型质量评估是蛋白质结构预测的重要组成部分。lDDT(local distance difference test)[50]作为一种评估蛋白质结构中所有原子的局部距离差异的分数,主要关注对应残基对的距离差异,因而不需要将候选结构与真实结构进行叠加,非常适合评估蛋白质的局部模型质量。第i个残差的lDDT评分和全局lDDT评分计算公式如下:
$$ \begin{array}{*{20}{c}} {{\rm{lDD}}{{\rm{T}}_i}{\rm{ = }}\dfrac{{4{p_1} + 3{p_2} + 2{p_3} + {p_4}}}{{4{p_0}}}}\\ {{\rm{lDD}}{{\rm{T}}_{{\rm{global}}}}{\rm{ = }}\dfrac{1}{L}\displaystyle\sum\limits_{i = 1}^L {l{\rm{DD}}{{\rm{T}}_i}} } \end{array} $$ 式中,p0是第i个残基和其他残基在15 Å以内距离的概率;p1是在15 Å以内第i个残基所有残基对的
$ {C_\beta }$ 距离偏差小于0.5 Å绝对值的概率。类似地,p2、p3和p4表示第i个残基在15 Å以内所有残基对${C_\beta } $ 距离偏差值分别为0.5–1.0Å、1.0–2.0Å和2.0–4.0Å绝对值的概率。在基于深度学习的模型质量评估方法中,特征设计和网络模型构建是影响评估性能的两个关键因素。文献[51]提出了一种基于超快速形状识别(ultrafast shape recognition, USR)的深度学习模型质量评估方法DeepUMQA。在深度残差网络的框架下,通过计算一组残基距离集合的一阶矩,引入残基级USR特征来描述残基与整体结构之间的拓扑关系,然后结合一维特征、二维特征和体素化特征来评估模型的质量。实验结果表明残基级的USR特征能与残基体素化特征形成互补,更全面地刻画残基的结构特性,显著提高了模型评估精度。在CASP13/14测试集以及CAMEO盲测结果显示,DeepUMQA及其改进版本多次在CAMEO周测中排名第一,性能优于大部分先进的模型质量评估方法。
-
为了真实反映近几年蛋白质结构预测方法的性能,根据最新的单域和多域结构预测相关论文进行了方法描述,并对论文中的实验结果进行性能分析与比较。
RocketX[52]是本课题组最新开发的基于深度学习几何约束预测及模型质量评估的从头蛋白质结构预测方法。构建了由残基间几何约束预测(GeomNet)、结构模拟和模型质量评估(EmaNet)组成的闭环反馈机制。在GeomNet中,从序列数据库中搜索的MSA中提取协同进化特征并送到改进的残差。
神经网络中,预测残基间的几何约束;在结构建模阶段利用预测的几何约束折叠结构模型;在EmaNet中,从折叠模型中提取一维和二维特征,通过深度残差神经网络估计残基间距离偏差和每残基lDDT,并将结果反馈给GeomNet作为动态特征来纠正几何约束预测以逐步提高模型精度。实验结果表明,闭环反馈机制显著提高了RocketX的性能,RocketX的预测精度优于方法trRosetta[16]和RaptorX[6, 53]。在CAMEO上的盲测结果显示,与集成了模板的先进方法相比,RocketX在Hard目标上具有一定优势。
表1给出了RocketX、trRosetta和RaptorX在483个非冗余基准测试蛋白上的平均预测结果。trRosetta的结果是从其官方服务器预测的,选择了“不使用模板”选项(
http://yanglab.nankai.edu.cn/trRosetta )。RaptorX的结果也是从其官方服务器(http://raptorx.uchicago.edu/ContactMap )预测的。表 1 RocketX、trRosetta和RaptorX在基准测试蛋白上的预测结果比较
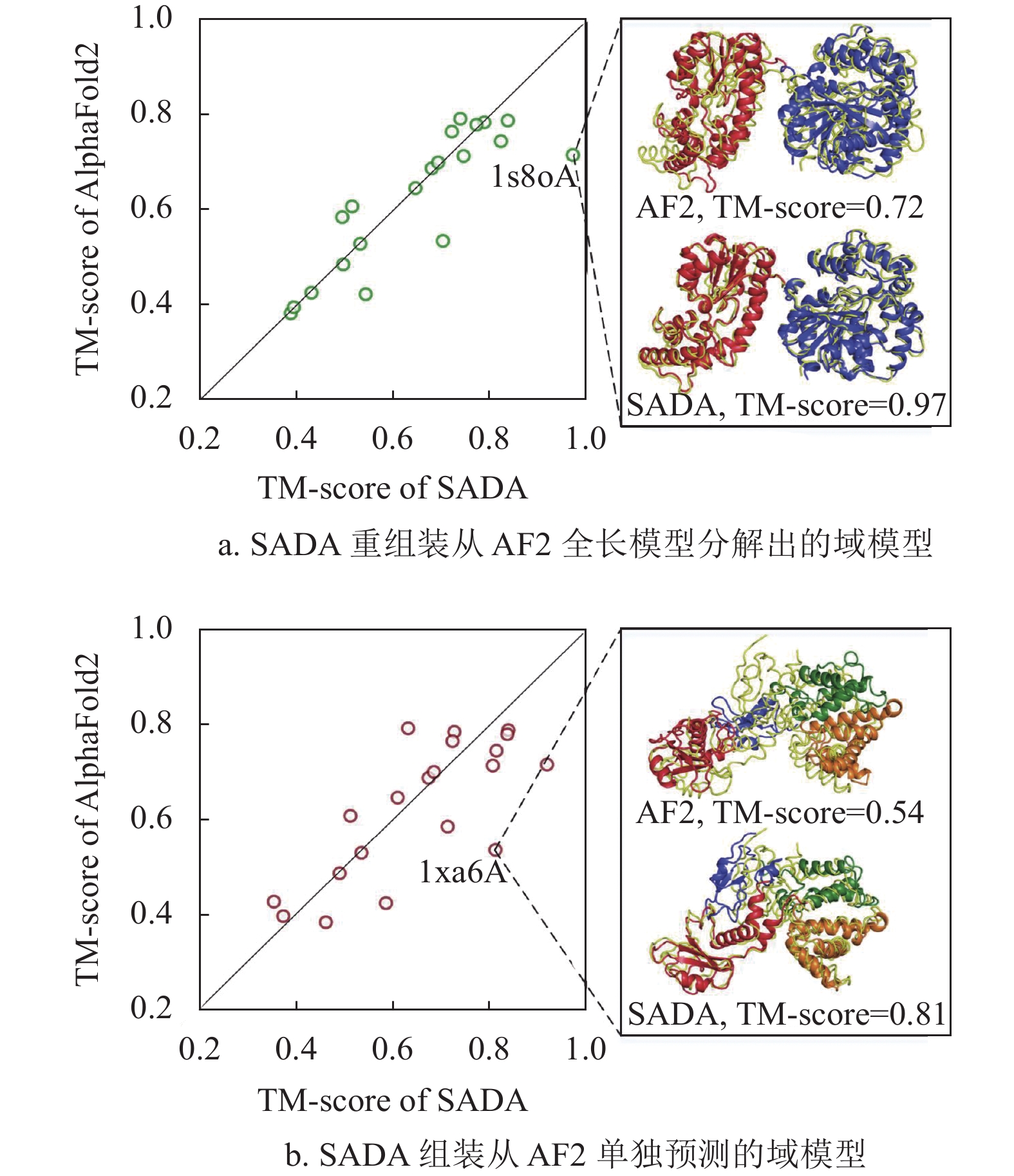
方法 RMSD TM-score #TM>0.5 #TM>0.9 RocketX 4.92 0.774 457 81 trRosetta 5.17 0.751 455 37 RaptorX 5.88 0.714 429 25 图2展示了SADA组装结构与AlphaFold2预测结构在20个人类多域蛋白上的对比实验结果。AlphaFold2的预测结构是从AlphaFold DB数据库中直接获取的。SADA分别组装了图2a从AlphaFold2的全链结构中拆分出来的单域模型和图2bAlphaFold2预测的单域模型。
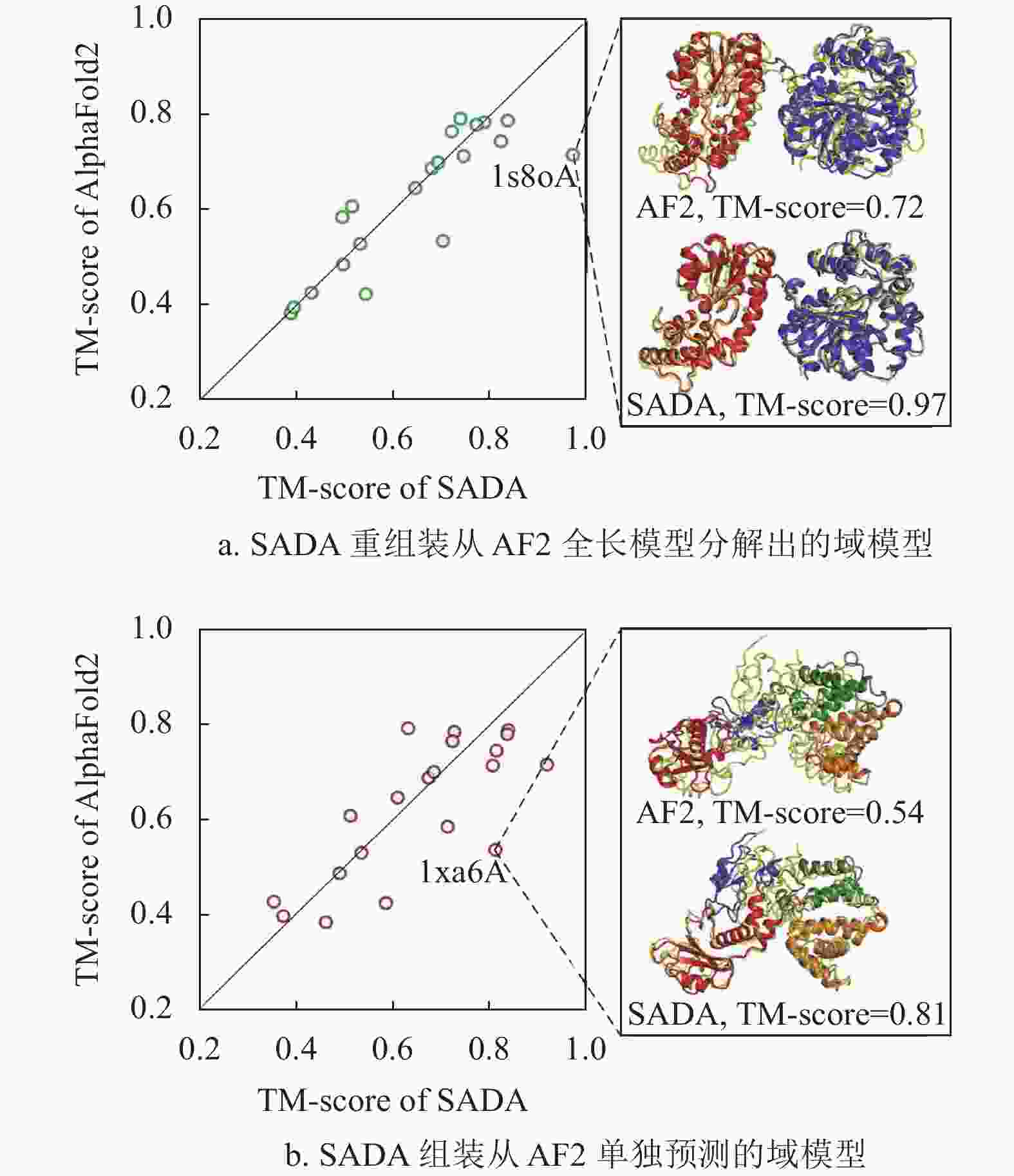
图 2 AlphaFold2的全链结构与SADA组装的全链结构之间的比较结果
SADA[54]是本课题组最新开发的一种由深度学习辅助的基于结构类似物的域组装方法。根据输入的单域模型,SADA首先从构建的多域蛋白质数据库MPDB中通过域级结构比对的方式找到输入域模型的全长结构类似物,并基于该结构类似物生成初始的全长模型。然后,利用蛋白质结构预测服务器RocketX中预测距离分布的几何约束网络模型GeomNet来预测全长模型的距离分布,并根据预测的距离分布和多域蛋白质的理化知识设计用于指导域组装的力场模型。最后,在力场模型的指导下,通过两阶段差分进化算法对初始模型进行域组装生成最终的全长模型。
-
自上世纪60年代以来,蛋白质结构预测问题一直是生物信息学关注的热点和难点问题。进入21世纪,尤其是在CASP系列赛事的推动下,在学术界和工业界的共同努力下,蛋白质结构预测领域取得了巨大突破。
在单域蛋白质预测方面,模板建模方法与无模板建模方法、物理化学能量模型和共进化的知识模型、基于片段组装的采样方法和几何优化方法的界限越来越模糊,他们之间相互补充,相互融合,共同促进。充分有效地利用蛋白质序列、宏基因组、结构数据将成为主流,深度学习模型从最初的接触、距离预测逐渐向方位、甚至是三维结构坐标方面发展。高通量预测的本源需求,使得预测方法从人工辅助方法逐渐向全自动化的方向发展。精度的提升,使得蛋白质结构预测技术和实验测定技术形成共存局面,即利用实验测定低分辨率结构辅助蛋白结构建模,反过来也利用预测技术提升实验测定精度和速度。模型质量评估技术将会成为预测技术进入实际应用的关键。
在多域蛋白质结构预测方面,随着单域结构预测取得的重大突破,预计在未来几年多域蛋白全长链建模将成为领域关注的热点问题。刚性组装会向柔性组装的方向发展,单域能量模型会向多域蛋白能量模型发展。基于共进化的几何特征(如接触、距离及方位)预测技术会向多域蛋白建模方向迁徙,多域蛋白结构预测方式从全长链建模会向结构域拆分、组装方式发展。在多域蛋白组装建模方式中,每个结构域的结构是已知的,序列比对、穿线比对方式会向结构比对的方式发展,开发高效的结构比对工具将成为一个重要的方向。柔性组装的要求使得需要高效率调整单结构域构象,这使得结构域的片段组装蒙特卡罗模拟方式向几何优化模拟方式发展。
An Overview of Multi-Domain Protein Structure Prediction Methods
-
摘要: 人工智能首次精确预测蛋白质三维结构入选《Science》杂志2020年十大科学突破,成为结构生物信息学领域的前沿方向。在自然界中,绝大多数单链蛋白中包含多个结构域。从生物学意义上来讲,结构域间缔结与协作对实现多个相关的功能至关重要。首先,介绍了蛋白质结构的预测技术发展及重要国际赛事CASP;其次,以单域蛋白结构预测方法、多域蛋白结构组装方法以及端到端的单体蛋白预测方法3部分对一些具有代表性的方法进行了简要阐述;然后,介绍了蛋白质结构预测研究中常用的数据库和模型质量评估指标,并比较了不同预测方法的性能;最后,分析总结了当前蛋白质结构预测方法的发展趋势,并对该领域未来的研究方向进行了展望。Abstract: Artificial intelligence accurately predicted the three-dimensional structure of proteins for the first time, which was selected as one of the top ten scientific breakthroughs in 2020 by "Science" magazine, and became a frontier direction in the field of structural bioinformatics. Most single-chain proteins in nature contain multiple domains. In a biological sense, inter-domain association and cooperation are crucial to achieve multiple related functions. This paper firstly introduces the development of protein structure prediction and the critical assessment of structure prediction (CASP); Secondly, some representative methods are briefly described in three parts: single-domain protein structure prediction methods, multi-domain protein structure assembly methods and end-to-end protein structure prediction methods; The commonly used databases and model quality evaluation indicators in protein structure prediction are then demonstrated, and the performances of the representative prediction methods are compared. Finally, we conclude with a brief overview of the future challenges and outstanding questions in the field.
-
表 1 RocketX、trRosetta和RaptorX在基准测试蛋白上的预测结果比较
方法 RMSD TM-score #TM>0.5 #TM>0.9 RocketX 4.92 0.774 457 81 trRosetta 5.17 0.751 455 37 RaptorX 5.88 0.714 429 25  下载: 导出CSV
下载: 导出CSV
-
[1] DOOLEY R. So much more to know…[J]. Science, 2005, 309(5731): 78-102. [2] KRYSHTAFOVYCH A, SCHWEDE T, TOPF M, et al. Critical assessment of methods of protein structure prediction (CASP)-Round XIII[J]. Proteins: Structure, Function, and Bioinformatics, 2019, 87(12): 1011-1020. doi: 10.1002/prot.25823 [3] LIU J, ZHOU X G, ZHANG Y, et al. CGLFold: A contact-assisted de novo protein structure prediction using global exploration and loop perturbation sampling algorithm[J]. Bioinformatics, 2020, 36(8): 2443-2450. doi: 10.1093/bioinformatics/btz943 [4] ALQURAISHI M. AlphaFold at CASP13[J]. Bioinformatics, 2019, 35(22): 4862-4865. doi: 10.1093/bioinformatics/btz422 [5] SHRESTHA R, FAJARDO E, GIL N, et al. Assessing the accuracy of contact predictions in CASP13[J]. Proteins: Structure, Function, and Bioinformatics, 2019, 87(12): 1058-1068. doi: 10.1002/prot.25819 [6] WANG S, SUN S Q, LI Z, et al. Accurate de novo prediction of protein contact map by ultra-deep learning model[J]. PLoS Computational Biology, 2017, 13(1): e1005324. doi: 10.1371/journal.pcbi.1005324 [7] XU J B, WANG S. Analysis of distance-based protein structure prediction by deep learning in CASP13[J]. Proteins: Structure, Function, and Bioinformatics, 2019, 87(12): 1069-1081. doi: 10.1002/prot.25810 [8] SENIOR A W, EVANS R, JUMPER J, et al. Improved protein structure prediction using potentials from deep learning[J]. Nature, 2020, 577(7792): 706-710. doi: 10.1038/s41586-019-1923-7 [9] JUMPER J, EVANS R, PRITZEL A, et al. Highly accurate protein structure prediction with AlphaFold[J]. Nature, 2021, 596(7873): 583-589. doi: 10.1038/s41586-021-03819-2 [10] CALLAWAY E. 'It will change everything': DeepMind's AI makes gigantic leap in solving protein structures[J]. Nature, 2020, 588(7837): 203-205. doi: 10.1038/d41586-020-03348-4 [11] BAEK M, DIMAIO F, ANISHCHENKO I, et al. Accurate prediction of protein structures and interactions using a three-track neural network[J]. Science, 2021, 373(6557): 871-876. doi: 10.1126/science.abj8754 [12] MOORE P B, HENDRICKSON W A, HENDERSON R, et al. The protein-folding problem: Not yet solved[J]. Science, 2022, 375(6580): 507. doi: 10.1126/science.abn9422 [13] CHOTHIA C, GOUGH J, VOGEL C, et al. Evolution of the protein repertoire[J]. Science, 2003, 300(5626): 1701-1703. doi: 10.1126/science.1085371 [14] ZHOU X G, HU J, ZHANG C X, et al. Assembling multidomain protein structures through analogous global structural alignments[J]. Proceedings of the National Academy of Sciences, 2019, 116(32): 15930-15938. doi: 10.1073/pnas.1905068116 [15] ZHENG W, LI Y, ZHANG C X, et al. Deep-learning contact-map guided protein structure prediction in CASP13[J]. Proteins: Structure, Function, and Bioinformatics, 2019, 87(12): 1149-1164. doi: 10.1002/prot.25792 [16] YANG J Y, ANISHCHENKO I, PARK H, et al. Improved protein structure prediction using predicted interresidue orientations[J]. Proceedings of the National Academy of Sciences, 2020, 117(3): 1496-1503. doi: 10.1073/pnas.1914677117 [17] ROHL C A, STRAUSS C E, MISURA K M, et al. Protein structure prediction using Rosetta[M]. [S.l.]: Elsevier, 2004. [18] 邓海游, 贾亚, 张阳. 蛋白质结构预测[J]. 物理学报, 2016, 65(17): 169-179. DENG H Y, JIA Y, ZHANG Y. Protein structure prediction[J]. Acta Phys Sin, 2016, 65(17): 169-179. [19] LI Z Q, SCHERAGA H A. Monte Carlo-minimization approach to the multiple-minima problem in protein folding[J]. Proceedings of the National Academy of Sciences, 1987, 84(19): 6611-6615. doi: 10.1073/pnas.84.19.6611 [20] KIHARA D, LU H, KOLINSKI A, et al. TOUCHSTONE: An ab initio protein structure prediction method that uses threading-based tertiary restraints[J]. Proceedings of the National Academy of Sciences, 2001, 98(18): 10125-10130. doi: 10.1073/pnas.181328398 [21] LINDORFF-LARSEN K, PIANA, DROR R O, et al. How fast-folding proteins fold[J]. Science, 2011, 334(6055): 517-520. doi: 10.1126/science.1208351 [22] ZHANG G J, MA L F, WANG X Q, et al. Secondary structure and contact guided differential evolution for protein structure prediction[J]. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 2018, 17(3): 1068-1081. [23] ZHOU X G, PENG C X, LIU J, et al. Underestimation-assisted global-local cooperative differential evolution and the application to protein structure prediction[J]. IEEE Transactions on Evolutionary Computation, 2019, 24(3): 536-550. [24] KUHLMAN B, BRADLEY P. Advances in protein structure prediction and design[J]. Nature Reviews Molecular Cell Biology, 2019, 20(11): 681-697. doi: 10.1038/s41580-019-0163-x [25] YANG J Y, ZHANG Y. I-TASSER server: New development for protein structure and function predictions[J]. Nucleic Acids Research, 2015, 43(W1): 174-181. doi: 10.1093/nar/gkv342 [26] YANG J Y, YAN R X, ROY A, et al. The I-TASSER Suite: Protein structure and function prediction[J]. Nature Methods, 2015, 12(1): 7-8. doi: 10.1038/nmeth.3213 [27] XU D, ZHANG Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field[J]. Proteins: Structure, Function, and Bioinformatics, 2012, 80(7): 1715-1735. doi: 10.1002/prot.24065 [28] ZHANG W X, YANG J Y, HE B J, et al. Integration of QUARK and I-TASSER for ab initio protein structure prediction in CASP11[J]. Proteins: Structure, Function, and Bioinformatics, 2016, 84: 76-86. doi: 10.1002/prot.24930 [29] HE B J, MORTUZA S, WANG Y T, et al. NeBcon: Protein contact map prediction using neural network training coupled with naïve Bayes classifiers[J]. Bioinformatics, 2017, 33(15): 2296-2306. doi: 10.1093/bioinformatics/btx164 [30] ZHANG C X, MORTUZA S, HE B J, et al. Template-based and free modeling of I-TASSER and QUARK pipelines using predicted contact maps in CASP12[J]. Proteins: Structure, Function, and Bioinformatics, 2018, 86: 136-151. doi: 10.1002/prot.25414 [31] LI Y, HU J, ZHANG C X, et al. ResPRE: High-accuracy protein contact prediction by coupling precision matrix with deep residual neural networks[J]. Bioinformatics, 2019, 35(22): 4647-4655. doi: 10.1093/bioinformatics/btz291 [32] ZHENG W, ZHANG C X, LI Y, et al. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations[J]. Cell Reports Methods, 2021, 1(3): 100014. doi: 10.1016/j.crmeth.2021.100014 [33] MORTUZA S, ZHENG W, ZHANG C X, et al. Improving fragment-based ab initio protein structure assembly using low-accuracy contact-map predictions[J]. Nature Communications, 2021, 12(1): 1-12. doi: 10.1038/s41467-020-20314-w [34] OVCHINNIKOV S, PARK H, VARGHESE N, et al. Protein structure determination using metagenome sequence data[J]. Science, 2017, 355(6322): 294-298. doi: 10.1126/science.aah4043 [35] ANISHCHENKO I, OVCHINNIKOV S, KAMISETTY H, et al. Origins of coevolution between residues distant in protein 3D structures[J]. Proceedings of the National Academy of Sciences, 2017, 114(34): 9122-9127. doi: 10.1073/pnas.1702664114 [36] MARKS D S, COLWELL L J, SHERIDAN R, et al. Protein 3D structure computed from evolutionary sequence variation[J]. PloS One, 2011, 6(12): e28766. doi: 10.1371/journal.pone.0028766 [37] EICKHOLT J, CHENG J L. Predicting protein residue-residue contacts using deep networks and boosting[J]. Bioinformatics, 2012, 28(23): 3066-3072. doi: 10.1093/bioinformatics/bts598 [38] 於东军, 李阳. 蛋白质残基接触图预测[J]. 南京理工大学学报:自然科学版, 2019, 43(1): 1-12. YU D J, LI Y. Protein residue-residue contact map prediction[J]. Journal of Nanjing University of Science and Technology, 2019, 43(1): 1-12. [39] HOU J, WU T Q, CAO R Z, et al. Protein tertiary structure modeling driven by deep learning and contact distance prediction in CASP13[J]. Proteins: Structure, Function, and Bioinformatics, 2019, 87(12): 1165-1178. doi: 10.1002/prot.25697 [40] SU H, WANG W K, DU Z Y, et al. Improved protein structure prediction using a new multi-scale network and homologous templates[J]. Advanced Science, 2021, 8(24): 2102592. doi: 10.1002/advs.202102592 [41] LI Y, ZHANG C X, BELL E W, et al. Ensembling multiple raw coevolutionary features with deep residual neural networks for contact-map prediction in CASP13[J]. Proteins: Structure, Function, and Bioinformatics, 2019, 87(12): 1082-1091. doi: 10.1002/prot.25798 [42] KANDATHIL S M, GREENER J G, JONES D T. Prediction of interresidue contacts with DeepMetaPSICOV in CASP13[J]. Proteins: Structure, Function, and Bioinformatics, 2019, 87(12): 1092-1099. doi: 10.1002/prot.25779 [43] MAO W Z, DING W Z, XING Y G, et al. AmoebaContact and GDFold as a pipeline for rapid de novo protein structure prediction[J]. Nature Machine Intelligence, 2020, 2(1): 25-33. doi: 10.1038/s42256-019-0130-4 [44] XU D, JAROSZEWSKI L, LI Z W, et al. AIDA: Ab initio domain assembly for automated multi-domain protein structure prediction and domain-domain interaction prediction[J]. Bioinformatics, 2015, 31(13): 2098-2105. doi: 10.1093/bioinformatics/btv092 [45] ZHANG Y, SKOLNICK J. TM-align: A protein structure alignment algorithm based on the TM-score[J]. Nucleic Acids Research, 2005, 33(7): 2302-2309. doi: 10.1093/nar/gki524 [46] JONES D T, THORNTON J M. The impact of AlphaFold2 one year on[J]. Nature Methods, 2022, 19(1): 15-20. doi: 10.1038/s41592-021-01365-3 [47] ORENGO C A, MICHIE A D, JONES S, et al. CATH-a hierarchic classification of protein domain structures[J]. Structure, 1997, 5(8): 1093-1109. doi: 10.1016/S0969-2126(97)00260-8 [48] CHANDONIA J M, FOX N K, BRENNER S E. SCOPe: Manual curation and artifact removal in the structural classification of proteins-extended database[J]. Journal of Molecular Biology, 2017, 429(3): 348-355. doi: 10.1016/j.jmb.2016.11.023 [49] PENG C X, ZHOU X G, XIA Y H, et al. MPDB: A unified multi-domain protein structure database integrating structural analogue detection[EB/OL]. [2021-10-28]. https://www.biorxiv.org/content/10.1101/2021.10.27.466092v1. [50] MARIANI V, BIASINI M, BARBATO A, et al. lDDT: A local superposition-free score for comparing protein structures and models using distance difference tests[J]. Bioinformatics, 2013, 29(21): 2722-2728. doi: 10.1093/bioinformatics/btt473 [51] GUO S S, LIU J, ZHOU X G, et al. DeepUMQA: Ultrafast shape recognition-based protein model quality assessment using deep learning[J]. Bioinformatics, 2022, 38(7): 1895-1903. doi: 10.1093/bioinformatics/btac056 [52] LIU J, HE G X, ZHAO K L, et al. De novo protein structure prediction by incremental inter-residue geometries prediction and model quality assessment using deep learning[EB/OL]. [2022-01-12]. https://www.biorxiv.org/content/10.1101/2022.01.11.475831v1. [53] XU J B. Distance-based protein folding powered by deep learning[J]. Proceedings of the National Academy of Sciences, 2019, 116(34): 16856-16865. doi: 10.1073/pnas.1821309116 [54] PENG C X, ZHOU X G, XIA Y H, et al. Structural analogue-based protein structure domain assembly assisted by deep learning[EB/OL]. [2022-03-08]. https://www.biorxiv.org/content/10.1101/2022.03.07.483151v1. -
 点击查看大图
点击查看大图
图(2) / 表(1)
计量
- 文章访问数: 5643
- HTML全文浏览量: 1748
- PDF下载量: 145
- 被引次数: 0



