 ISSN
ISSN 


-
RNA可变剪接(alternative splicing, AS)是真核生物中剪接体选择性剪切外显子形成不同RNA异构体的过程[1],是调节基因表达、产生蛋白质分子多样性的关键环节[2]。文献[3-4]发现,90%以上的人类基因都会经历可变剪接。可变剪接不仅增加了生物分子的复杂性、多样性,还与疾病的发生有关[5]。如癌基因ETS中外显子7b的剪接与细胞增殖的减少有关[6]。由于剪接因子MBNL3的调节,lncRNA PXN-AS1的外显子4被保留在转录本中,促进了肝癌的发生[7]。丙酮酸激酶前体mRNA在剪接过程中保留了外显子10,产生的亚型PKM2过度表达,导致了肿瘤的发生[8]。可变剪接产生的雌激素受体α和β的变体ERα46和ERβ1则与乳腺癌密切相关[9]。因此,对可变剪接调控机制的研究尤为重要。
RNA可变剪接并非独立的生物过程,而是与转录过程存在着时空上的偶联[10]。除了位于外显子和内含子中的顺式和反式元件[11-12],可变剪接还受到组蛋白修饰、DNA甲基化等表观遗传因素的调节。随着DNA元件百科全书(encylopedia of DNA elements, ENCODE)计划的深入开展,组蛋白修饰参与可变剪接调控的现象也被逐渐发现,尤其是出现在内含子/外显子区的组蛋白甲基化和组蛋白乙酰化修饰与细胞系特异性可变剪接密切相关[13]。组蛋白乙酰化修饰对RNA可变剪接的调控在神经细胞粘附分子(neural cell adhesion molecule, NCAM)基因中被发现。NCAM基因中盒式外显子的切除与外显子中高含量的H3K9ac密切相关[14]。NCAM基因盒式外显子中H3K9ac改变了该区域的染色质结构,造成转录过程中RNA Pol II 的移动速率加快,从而导致NCAM基因中盒式外显子发生了可变剪接。在小鼠胚胎干细胞分化为神经元的过程中,组蛋白乙酰化修饰还调控了Nf1基因可变外显子23a和Fas基因可变外显子6的可变剪接[15]。另外,成纤维原细胞生长因子受体基因(fibroblast growth factor receptor2, FGFR2)中外显子区的H3K36me3是可变剪接调控蛋白的识别标记。FGFR2基因中存在一对互斥外显子IIIb和IIIc,FGFR2-IIIb只在上皮细胞中表达,而FGFR2-IIIc却在间质细胞中表达[16]。通过分析间质细胞和上皮细胞中FGFR2基因的组蛋白修饰后发现,与上皮细胞相比,间质细胞的FGFR2基因外显子中富含H3K36me3。因此,染色质重塑复合物MRG15通过与FGFR2基因中的H3K36me3相互作用,能够招募多聚嘧啶结合蛋白(polypyrimidine tract-binding protein, PTB)与FGFR2-IIIb外显子侧翼的内含子剪接抑制子结合,从而使得FGFR2-IIIb在间质细胞中被切除。
组蛋白修饰间还存在因果关联,多种组蛋白修饰组合在一起形成级联,共同调控基因表达[17-18]。在可变剪接过程中,不同类型的组蛋白修饰可以通过协同或拮抗方式调控剪接复合因子的招募,从而实现对RNA剪接过程的调控。文献[19]发现,人胚肺成纤维细胞系(IMR90 cell line)中BIN1基因的可变剪接就是多种组蛋白修饰(H3K36me3、H3K4me3、H2BK12ac、H4K5ac)协同作用的结果。这些工作既为研究组蛋白甲基化和组蛋白乙酰化修饰调控可变剪接提供了理论依据,又显示出从组蛋白修饰等表观遗传因素中挖掘新信息是认识可变剪接调控机制的新途径。
外显子跳跃模式是哺乳动物最常见的可变剪接模式[20-21]。文献[22-23]发现了CD4+ T细胞外显子跳跃模式中多种组蛋白修饰在外显子和内含子中富集程度的差异性,并利用组蛋白修饰差异信息对包含和排除外显子进行了识别。通过构建组蛋白修饰间的相互作用网络,文献[24]还分析了CD4+ T细胞的外显子跳跃模式中组蛋白修饰之间的因果关系。组蛋白修饰不仅可以通过改变RNA Pol II的延伸率或招募剪接因子参与可变剪接的调节[25],还能通过彼此之间的相互作用调控可变剪接。通过对人胚胎干细胞系(H1 cell line)的转录组学和表观遗传组学数据进行关联分析,文献[26]发现组蛋白修饰的动态变化与细胞特异性剪接机制相关。最近,文献[27]发现了组蛋白修饰在H1和IMR90细胞系的外显子跳跃模式中的协同分布规律,并在MCF10a、K562和HeLa等细胞系中进行了验证,遗憾的是其并未阐明组蛋白修饰间的因果关系。
鉴于此,本文以IMR90细胞系中外显子跳跃剪接事件为研究对象,分析了28种组蛋白修饰在外显子跳跃模式排除和包含外显子上的相关性,通过构建贝叶斯网络推断了组蛋白修饰间的因果关系。
-
人类基因组(GRCh37版本) cDNA序列和基因注释文件来自Ensembl数据库(
https://asia.ensembl.org/index.html )。IMR90细胞系转录组测序数据(RNA-seq)来自GEO数据库(https://www.ncbi.nlm.nih.gov/geo/ , GSM2400222)。使用高通量数据过滤工具Trimmomatic[28](版本0.39-2)对RNA-seq进行处理,参数设置为“PEILLUMINACLIP: TruSeq3-PE-2.fa:2:30:10:1:true LEADING:3 TRAILING:3 SLIDINGWINDOW:4:20 MINLEN:40 TOPHRED33”。从GEO数据库(登录号GSE16256)获取IMR90细胞系的28种组蛋白修饰数据的bed文件,相应的GEO样本号如表1所示。
表 1 组蛋白修饰的GEO样本号
组蛋白修饰 GEO样本号 H2A.Z GSM818007 H2AK5ac GSM521866 H2AK9ac GSM818012 H2BK120ac GSM521869 H2BK12ac GSM521871 H2BK15ac GSM521875 H2BK20ac GSM521879 H2BK5ac GSM818017 H3K14ac GSM521881 H3K18ac GSM469965 H3K23ac GSM521885 H3K27ac GSM469966 H3K27me3 GSM469968 H3K36me3 GSM521890 H3K4ac GSM521893 H3K4me1 GSM521895 H3K4me2 GSM521899 H3K4me3 GSM469970 H3K56ac GSM521902 H3K79me1 GSM521904 H3K79me2 GSM521909 H3K9ac GSM469973 H3K9me1 GSM752986 H3K9me3 GSM469974 H4K20me1 GSM521915 H4K5ac GSM469975 H4K8ac GSM521919 H4K91ac GSM521924 -
统计RNA-seq比对到基因组上的短读序列(reads)的数量,即每个转录本的表达量。由于测序深度、转录本长度等因素的影响,表达量并不能直接代表转录本的相对丰度。因此对这些表达量进行TPM[29]标准化:
$$ {\rm{TPM}}=\frac{{N}_{i}/{L}_{i} \times {10}^{6}}{{\rm{sum}}({N}_{1}/{L}_{1}+{N}_{2}/{L}_{2}+\dots +{N}_{n}/{L}_{n})} $$ (1) 式中,Ni表示映射到第i个转录本的reads数;Li表示第i个转录本的长度。
使用Salmon[30](版本1.5.1)计算转录本的TPM值,首先在参考cDNA序列上建立索引,参数使用默认值。生成的索引与处理后的RNA-seq进行定量,参数设置为“-l A–validateMappings–gcBias--seqBias”。
-
外显子跳跃事件中被保留在成熟转录本中的外显子为包含外显子,被剪接的外显子为排除外显子。根据发生外显子跳跃事件的基因上所有转录本的TPM,计算每个外显子的包含率(percent spliced in, PSI):
$$ {\rm{PSI}}=\frac{\displaystyle\sum _{i\in {S}_{1}}{{\rm{TPM}}}_{i}}{\displaystyle\sum _{j\in {S}_{1}\cup {S}_{2}}{{\rm{TPM}}}_{j}} $$ (2) 式中,TPMi表示基因的第i个包含跳跃外显子的转录本的TPM值;S1表示包含跳跃外显子的转录本集合;TPMj表示基因的第j个转录本的TPM值;S2表示不包含跳跃外显子的转录本集合。PSI的值越大,表示外显子被包含在最终转录本的概率越大,PSI的值越小,外显子被排除的概率越大。
使用Suppa软件[31](版本2.3),根据基因注释信息生成外显子跳跃事件,并利用TPM计算每个跳跃外显子的PSI,所有参数使用默认值。最终获得36468个外显子跳跃事件,其外显子PSI值的范围为0~1。PSI为1或0时,表示外显子被包含或被排除在基因的所有转录本中。为了适当扩充数据量,定义PSI>0.85的外显子为包含外显子,PSI<0.15的外显子为排除外显子。由于外显子过短无法匹配到组蛋白修饰数据,因此只保留长度在150~300 bp之间的外显子,最后获得5122个包含外显子和4638个排除外显子。
-
根据bed文件中组蛋白修饰在基因组上的位点信息,使用BEDTools[32](版本2.30.0)计算其在排除和包含外显子上的富集程度(reads数)。然后根据reads数,使用R语言(版本4.1.2)Hmisc包中的rcorr函数分别计算排除和包含外显子上组蛋白修饰之间的皮尔逊相关系数。由此获得28种组蛋白修饰间的相关性系数矩阵,用corrplot绘制相关性热图,并设置统计显著性阈值p=0.05。
-
贝叶斯网络是一种描述变量间因果关系的统计推理模型,其网络拓扑结构是有向无环图(directed acyclic graph, DAG)[33]。网络中包括节点和边,节点表示随机变量,边表示变量之间的条件依赖关系。所有边为单向箭头,箭头指向的节点为子节点,箭头另一端的节点为父节点。在贝叶斯网络中,如果两个节点之间不存在有向边,则说明这两个节点彼此条件独立;如果两个节点间存在有向边,则说明这两个节点间存在因果关系。
根据1.4节的reads数,对排除和包含外显子上组蛋白修饰进行离散化,“1”表示外显子上存在组蛋白修饰(reads>1),“0”表示外显子上不存在组蛋白修饰(reads=0)。为了得到稳定的组蛋白修饰相互作用网络,采用10交叉检验法对所得网络进行验证。首先将组蛋白修饰数据平均分成10份,其中9份被用作训练集,用于构建基本的贝叶斯网络,1份被用作测试集对网络的稳定性进行验证。使用WinMine(
https://www.microsoft.com/en-us/research/project/winmine-toolkit/ )构建组蛋白修饰的贝叶斯网络,最终得到10个不同的网络。如果连接节点的有向边在10个网络中均存在,则将这些节点和边保留,用以构建最终网络。利用Cytoscape[34](版本3.8.2)软件展示所得的网络拓扑结构。网络中节点表示组蛋白修饰,并根据1.4节得到的相关系数对边赋值。 -
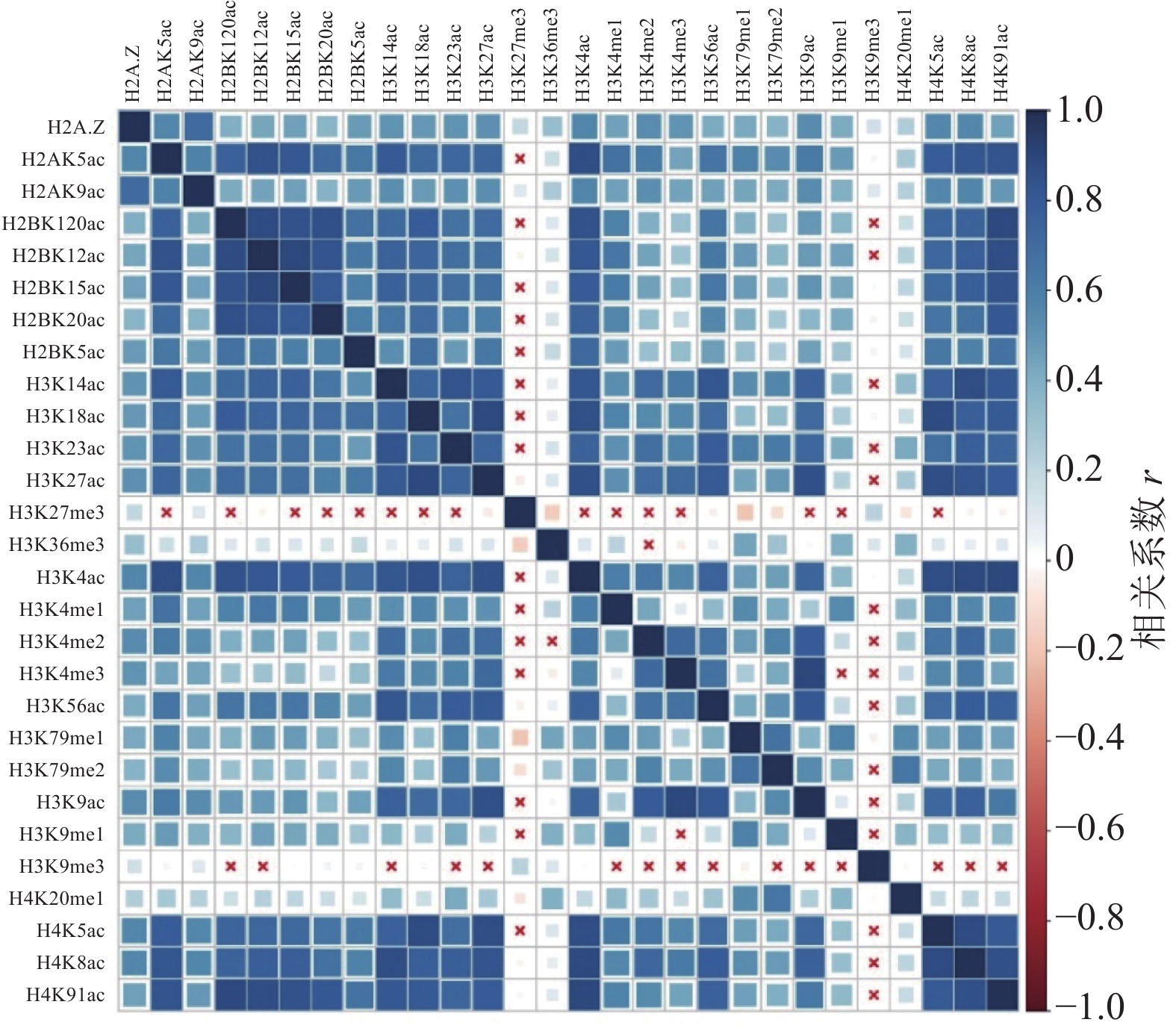
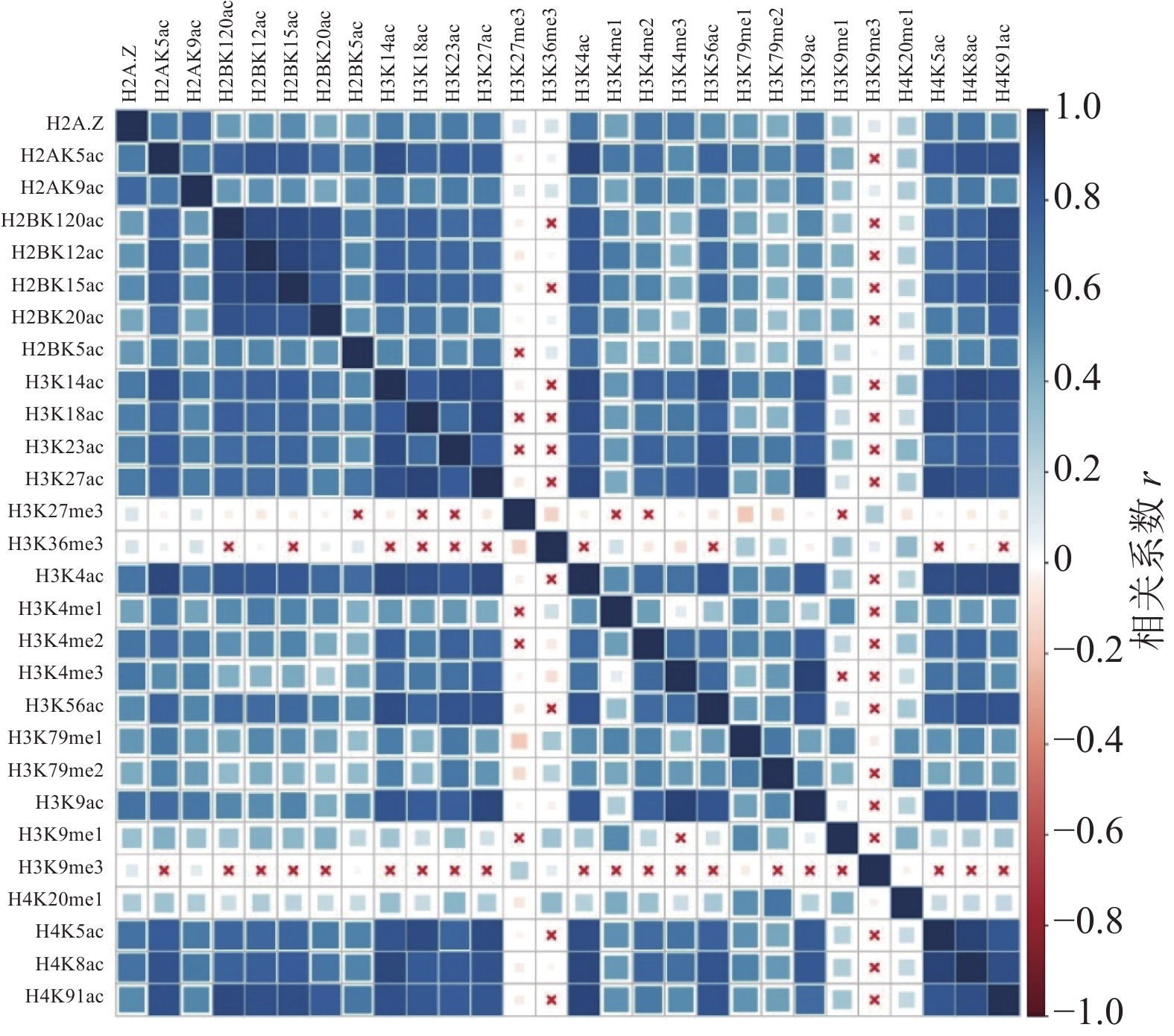
计算组蛋白修饰之间的相关系数,通过绘制相关性热图,分析组蛋白修饰间的相关性,如图1和图2所示。图中“×”表示组蛋白修饰间的相关性不显著,色块大小表示相关性强弱。结果表明,IMR90细胞系中排除和包含外显子上大部分组蛋白修饰之间存在协同(r>0)或拮抗作用(r<0)。
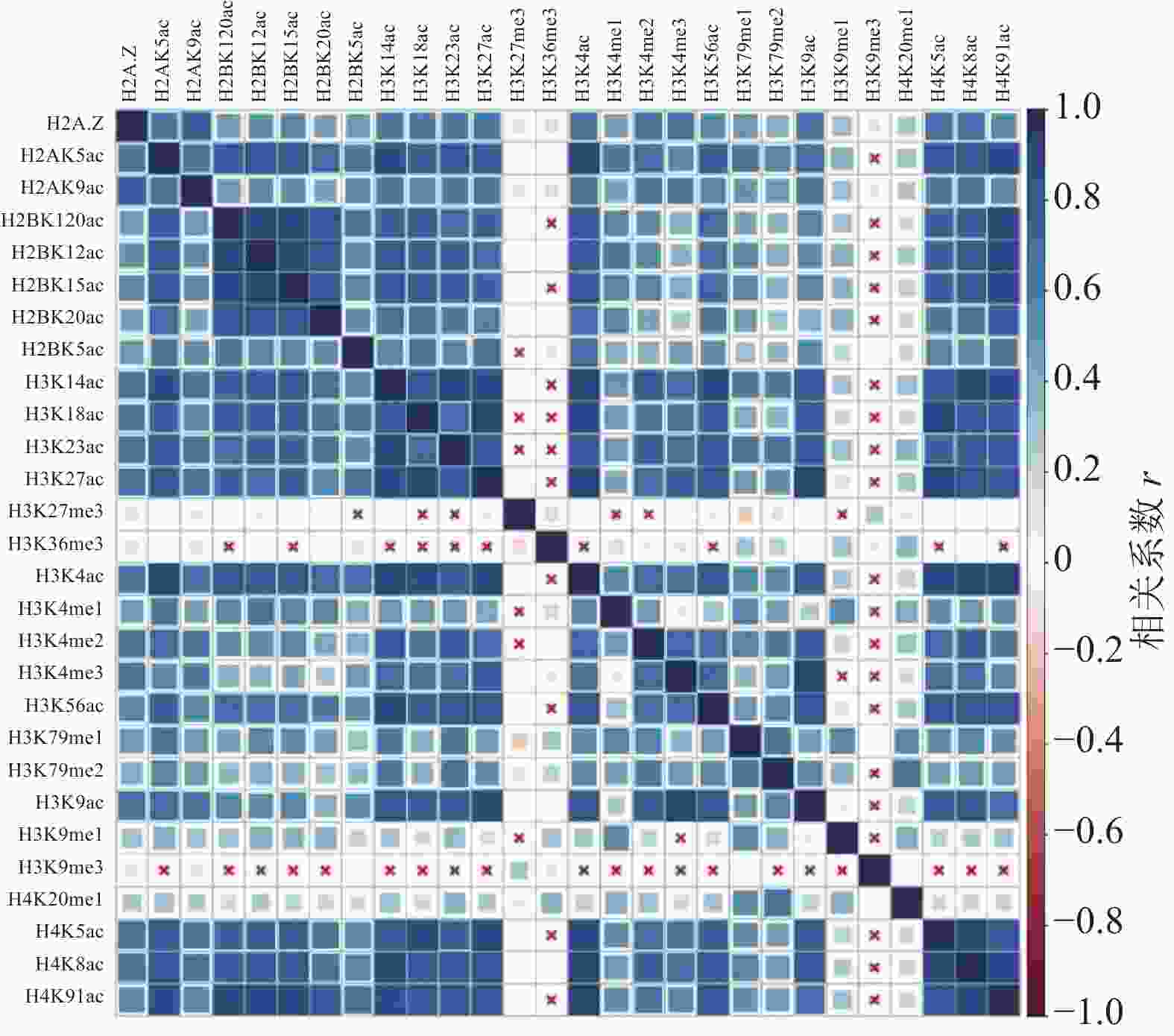
图 1 排除外显子上组蛋白修饰之间的相关性
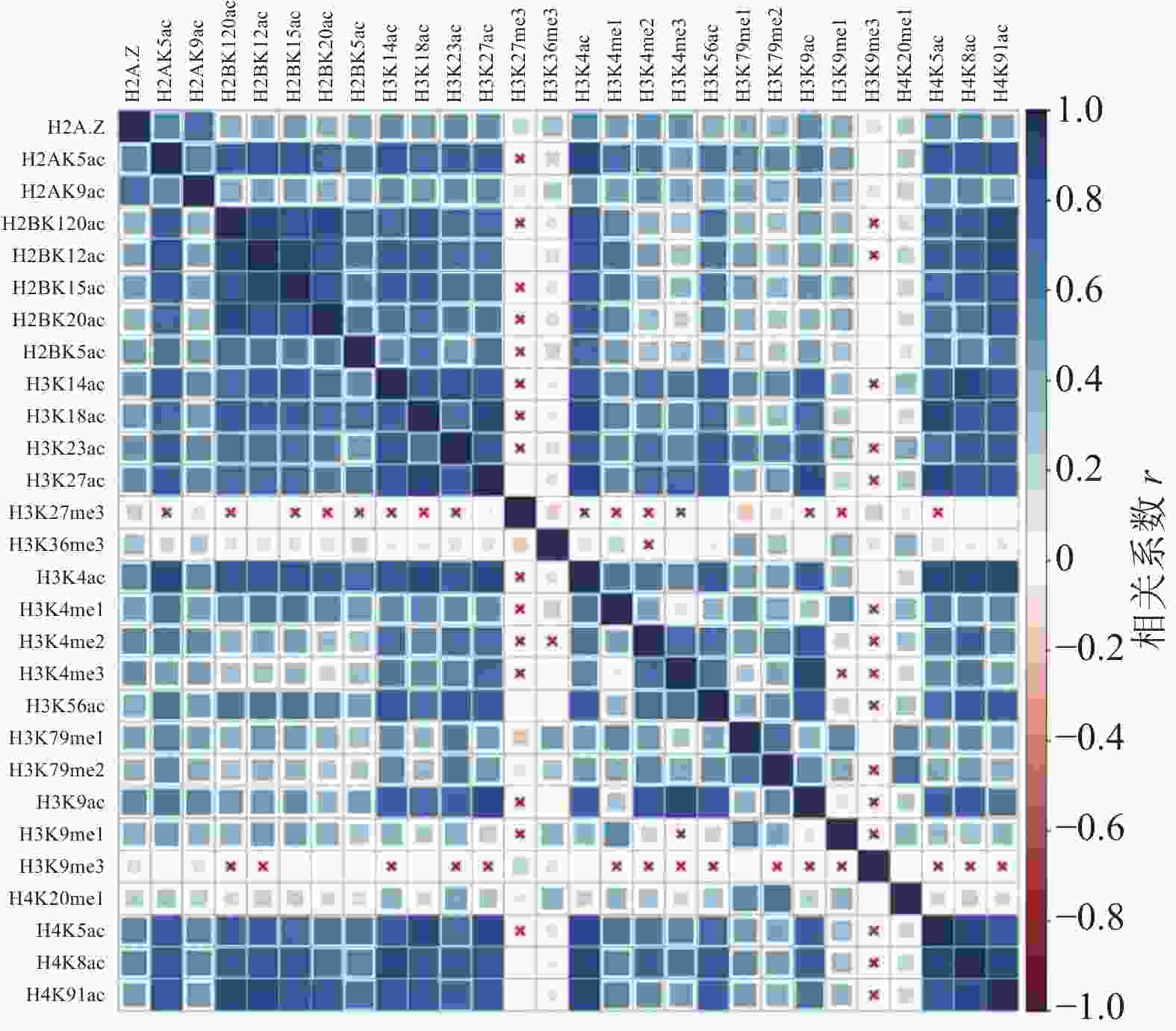
图 2 包含外显子上的组蛋白修饰之间的相关性
具体而言,组蛋白乙酰化之间表现出了正相关性,并且具有强正相关的组蛋白修饰存在于同一组蛋白上(H2B:H2BK120ac、H2BK12ac、H2BK15ac、H2BK20ac;H3:H3K14ac、H3K18ac、H3K23ac、H3K27ac;H4:H4K5ac、H4H8ac、H4K91ac)。在CD4+T细胞中也发现了组蛋白H3和H4上相同的组合模式,但其正相关性弱于IMR90细胞系[24]。
组蛋白甲基化之间同时表现出正相关性和负相关性,如H4K20me1和H3K79me1、H3K79me2之间正相关,而H3K27me3和H3K36me3、H3K79me1、H3K79me2之间负相关。在间质细胞中存在相同的现象,H3K27me3和H3K36me3对基因FGFR2的外显子Ⅲb的保留和剪切表现出了拮抗作用[35]。
排除和包含外显子上组蛋白修饰间的相关性也存在差异。如H3K36me3在包含外显子上与10种组蛋白修饰(H2BK120ac、H2BK15ac、H3K14ac、H3K18ac、H3K23ac、H3K27ac、H3K4ac、H3K56ac、H4K5ac、H4K91ac)之间正相关,如图2所示,而在排除外显子中则未发现显著的相关性,如图1所示。文献[19, 36]研究发现,H3K36me3富集在包含外显子上,推测其可能与不同的组蛋白修饰形成组合模式,参与了RNA可变剪接的调控。
-
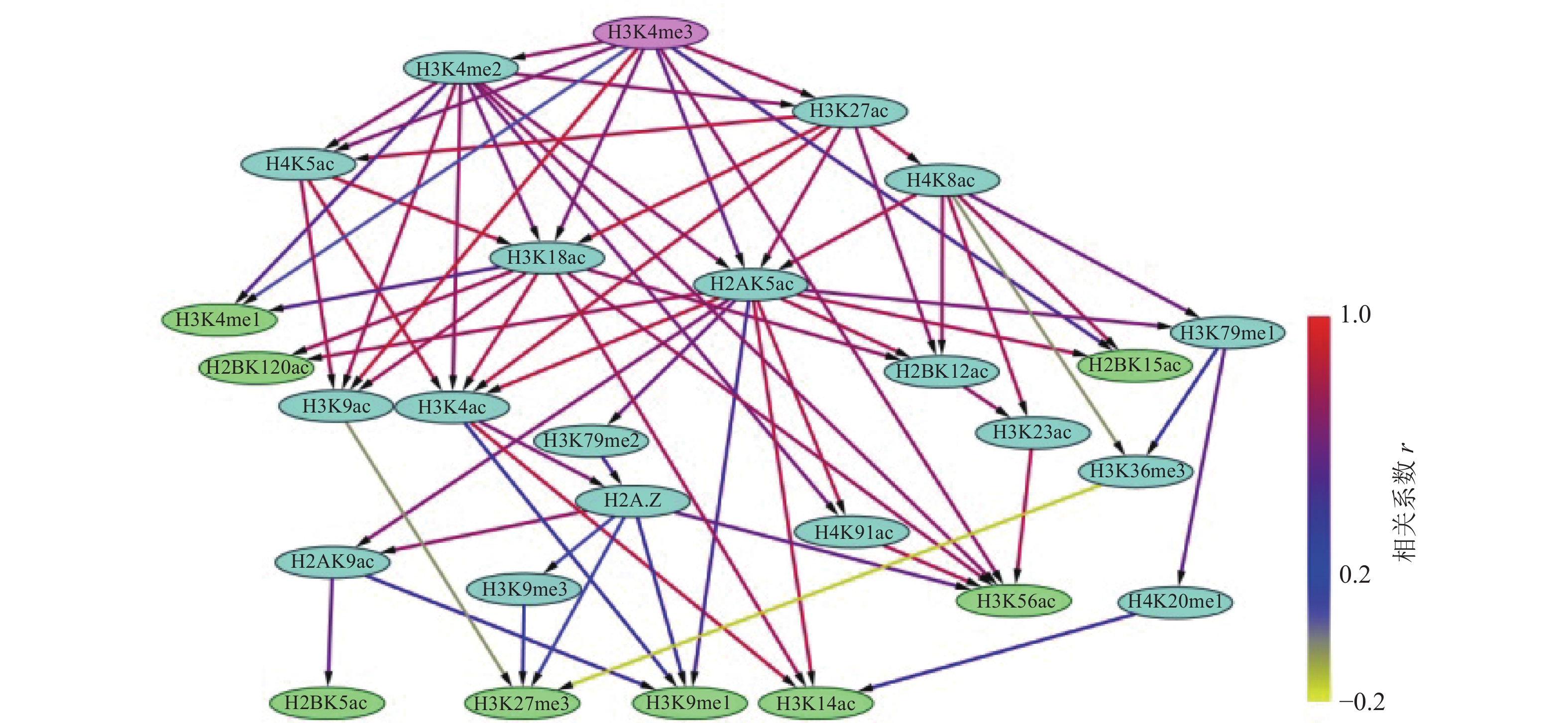
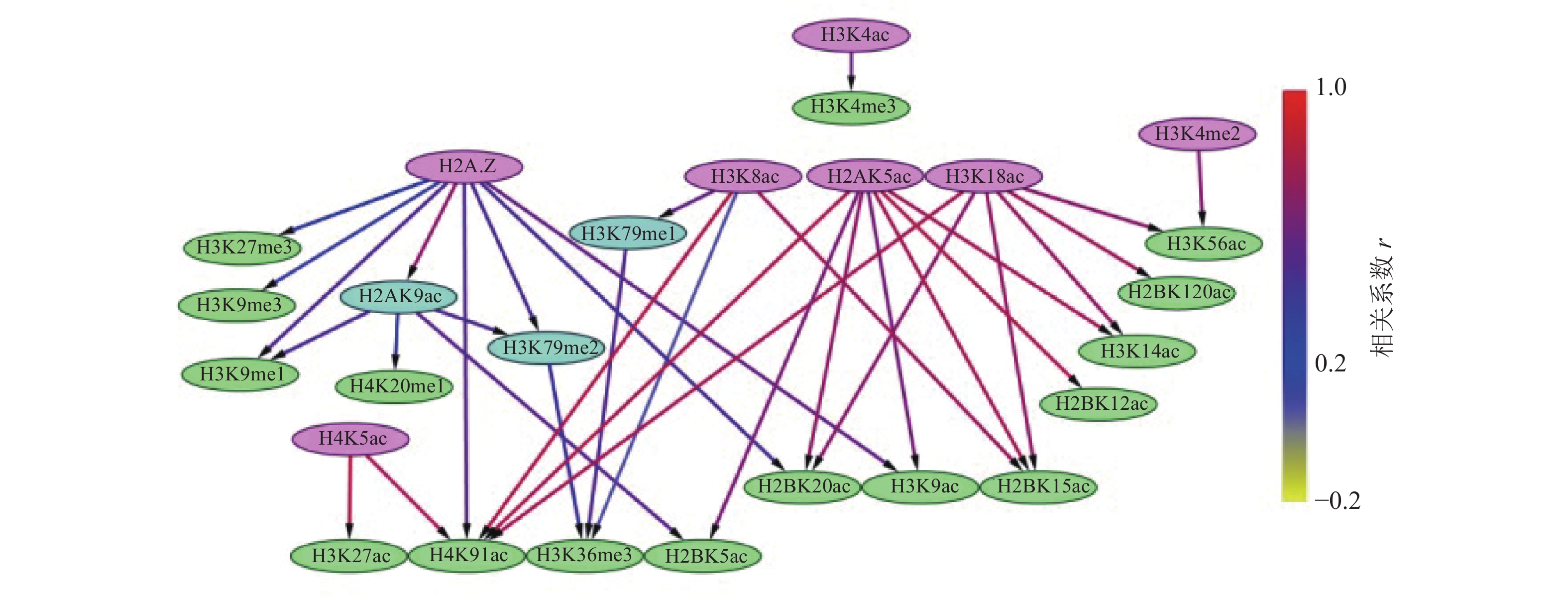
通过构建IMR90细胞中组蛋白修饰间的贝叶斯网络,如图3和4所示,对外显子跳跃剪接事件中组蛋白修饰间的因果关系进行推断,发现贝叶斯网络中涉及的组蛋白修饰不仅包括同一组蛋白中同一氨基酸不同程度的修饰,还包括不同组蛋白中同一类型的组蛋白修饰,以及不同组蛋白中不同类型的组蛋白修饰。
在网络拓扑结构图中,只有子节点的组蛋白修饰用紫色标出;只有父节点的组蛋白修饰用绿色标出;既有父节点也有子节点的组蛋白修饰用蓝色标出。
比较两组网络拓扑结构发现,排除和包含外显子上组蛋白修饰之间存在18种相同的因果关系,分别为:H2A.Z→H2AK9ac→H2BK5ac、H2AK9ac→H3K9me1、H2A.Z→H3K27me3、H2A.Z→H3K9me1、H2A.Z→H3K9me3、H2AK5ac→H2BK12ac、H2AK5ac→H3K14ac、H2AK5ac→H2BK15ac、H2AK5ac→H4K91ac、H3K18ac→H3K14ac、H3K18ac→H2BK120ac、H3K18ac→H3K56ac、H3K4me2→H3K56ac、H4K8ac→H3K79me1→H3K36me3、H4K8ac→H3K36me3、H4K8ac→H2BK15ac。
在排除外显子中,27种组蛋白修饰构成了71种因果关系,如图3所示。H3K4me3直接或间接调控了剩余的26种组蛋白修饰。8种组蛋白修饰(H2BK120ac、H2BK15ac、H2BK5ac、H3K14ac、H3K27me3、H3K4me1、H3K56ac、H3K9me1)可能直接与剪接因子相互作用,参与了可变剪接调控。在包含外显子中,26种组蛋白修饰构成了35种因果关系,如图4所示,其中7种组蛋白修饰(H2A.Z、H2AK5ac、H3K18ac、H3K4ac、H3K4me2、H4K5ac、H4K8ac)直接或间接调节剩余的19种组蛋白修饰。16种组蛋白修饰(H2BK120ac、H2BK12ac、H2BK15ac、H2BK20ac、H2BK5ac、H3K14ac、H3K27ac、H3K27me3、H3K36me3、H3K4me3、H3K56ac、H3K9ac、H3K9me1、H3K9me3、H4K20me1、H4K91ac)可能直接与剪接因子相互作用,参与了可变剪接的调控。
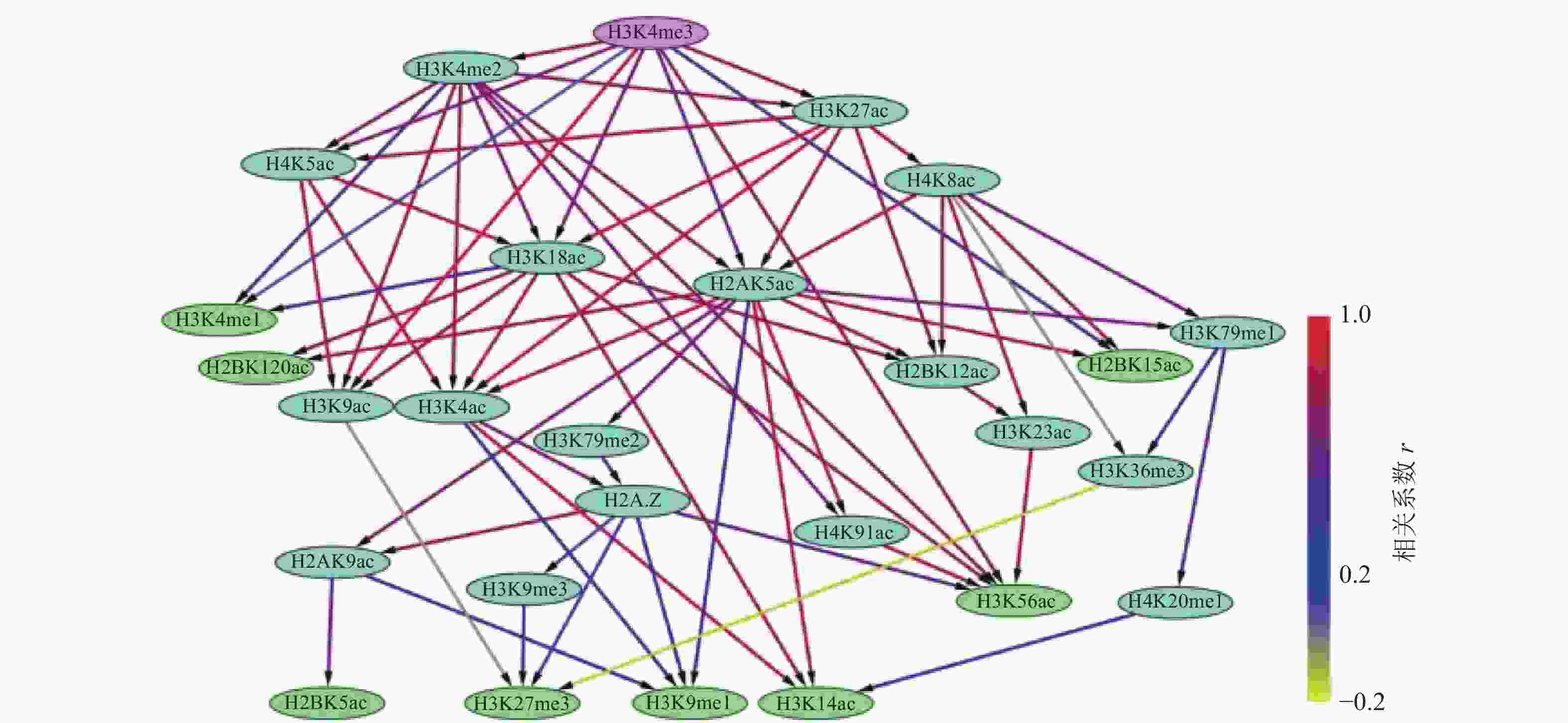
图 3 排除外显子上组蛋白修饰之间的贝叶斯网络
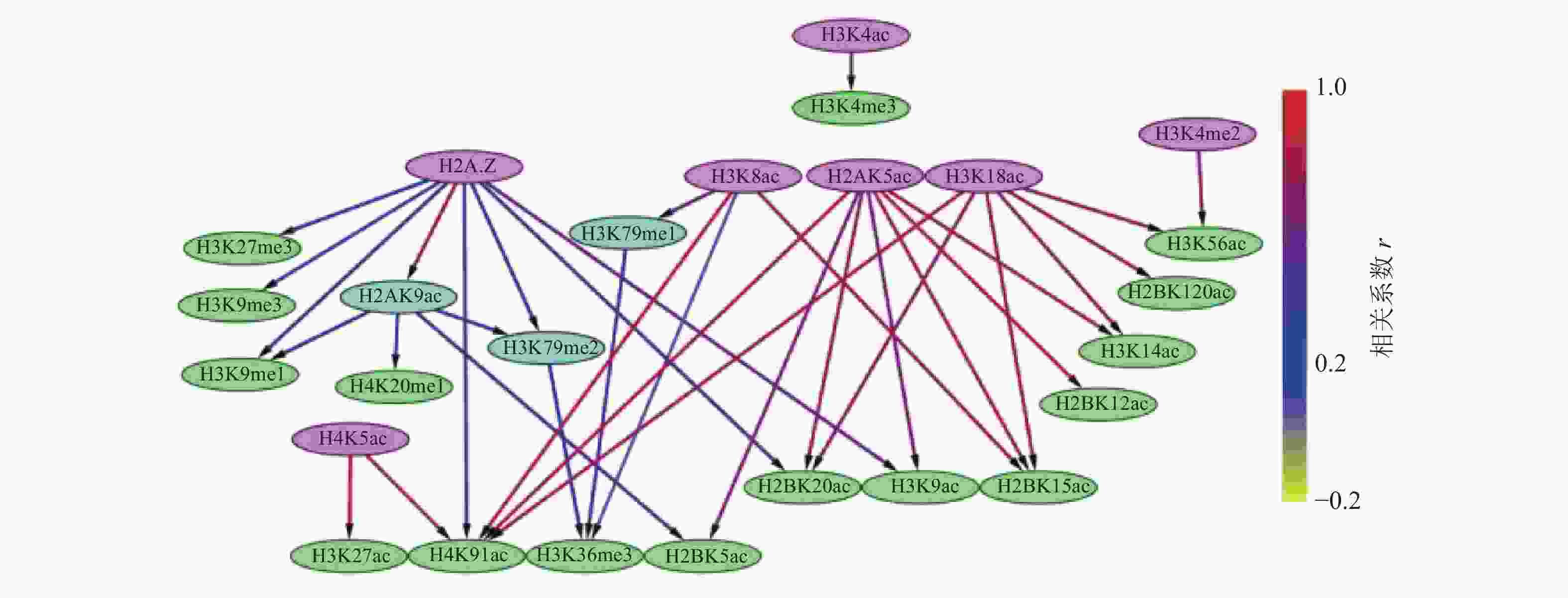
图 4 包含外显子上组蛋白修饰之间的贝叶斯网网络
此外,排除和包含外显子对应的网络复杂程度也存在明显差异。排除外显子对应的网络拓扑结构复杂,组蛋白修饰组合的调控路径长,并且部分组蛋白修饰间表现出了拮抗关系,如H4K8ac→H3K36me3→H3K27me3、H3K9ac→H3K27me3,如图3所示。包含外显子对应的网络拓扑结构则相对简单,组蛋白修饰组合的调控路径较短,如图4所示。另外IMR90细胞中排除和包含外显子上的H3K27ac和H4K5ac之间因果关系相反。
组蛋白修饰在可变剪接过程中的调控作用已被发现,如H3K4me3与U2 snRNP结合可调节剪接速率[37-38]。IMR90细胞系包含外显子上的调控网络显示H3K4me3可能受到了H3K4ac的影响,如图4所示。而排除外显子上H3K4me3不仅与9种组蛋白修饰(H3K4me2、H4K5ac、H3K4me1、H3K9ac、H3K18ac、H2AK5ac、H3K56ac、H2BK15ac、H3K27ac)之间存在直接因果关系,还存在于所有的调控路径中,如图3所示。在间质细胞中,H3K36的去甲基化酶KDM2a被募集到富含H3K27me3的区域,保持了低H3K36me3水平,从而促进了外显子Ⅲb的包含[35]。这一结果表明,H3K36me3与H3K27me3之间的拮抗作用调控了可变剪接。有意思的是,在IMR90细胞系的排除外显子中也发现了H3K36me3和H3K27me3之间的拮抗关系,如图3所示。由此推测,IMR90细胞系中的可变剪接受到了组蛋白修饰间相互作用的调控,相关结果还需进一步实验验证。
-
本文通过对IMR90细胞系中组蛋白修饰间的相关性进行分析,发现了外显子跳跃剪接事件中组蛋白修饰间存在明显的组合模式。通过构建贝叶斯网络,分析了排除和包含外显子中组蛋白修饰间的因果关系。由此推测,IMR90细胞系排除和包含外显子的组蛋白修饰可能通过组合方式与剪接因子相互作用,直接或间接地参与了可变剪接的调控。
Analysis of Combinatorial Pattern of Histone Modifications in Exon Skipping Event
-
摘要: 可变剪接是基因表达调控的重要过程。组蛋白修饰参与了可变剪接,但具体调控机制尚不清楚。为了研究组蛋白修饰与可变剪接的关系,首先基于人胚肺成纤维细胞系的RNA-Seq数据,获得了该细胞系外显子跳跃模式的可变剪接数据。在此基础上,分析了外显子跳跃模式中排除和包含外显子中组蛋白修饰间的相关性。通过构建组蛋白修饰间的贝叶斯网络,推断了人胚肺成纤维细胞系外显子跳跃可变剪接事件中组蛋白修饰之间的因果关系。Abstract: Alternative splicing is a key process of gene expression regulation. The histone modifications are closely correlated with alternative splicing, however, its regulatory mechanisms remain unclear. In order to reveal the relationships between histone modifications and alternative splicing, by analyzing the RNA-Seq data, the exon skipping events from human embryonic lung fibroblast cell line were obtained. Subsequently, the relationships among different kinds of histone modifications were analyzed for both excluded and included exons in exon skipping event. Finally, the Bayesian network was built to deduce the casual relationships among histone modifications in exon skipping event.
-
Key words:
- alternative splicing /
- Bayesian network /
- exon skipping event /
- histone modifications
-
表 1 组蛋白修饰的GEO样本号
组蛋白修饰 GEO样本号 H2A.Z GSM818007 H2AK5ac GSM521866 H2AK9ac GSM818012 H2BK120ac GSM521869 H2BK12ac GSM521871 H2BK15ac GSM521875 H2BK20ac GSM521879 H2BK5ac GSM818017 H3K14ac GSM521881 H3K18ac GSM469965 H3K23ac GSM521885 H3K27ac GSM469966 H3K27me3 GSM469968 H3K36me3 GSM521890 H3K4ac GSM521893 H3K4me1 GSM521895 H3K4me2 GSM521899 H3K4me3 GSM469970 H3K56ac GSM521902 H3K79me1 GSM521904 H3K79me2 GSM521909 H3K9ac GSM469973 H3K9me1 GSM752986 H3K9me3 GSM469974 H4K20me1 GSM521915 H4K5ac GSM469975 H4K8ac GSM521919 H4K91ac GSM521924  下载: 导出CSV
下载: 导出CSV
-
[1] LEE Y, RIO D C. Mechanisms and regulation of alternative pre-mRNA splicing[J]. Annu Rev Biochem, 2015, 84: 291-323. doi: 10.1146/annurev-biochem-060614-034316 [2] KELEMEN O, CONVERTINI P, ZHANG Z, et al. Function of alternative splicing[J]. Gene, 2013, 514(1): 1-30. doi: 10.1016/j.gene.2012.07.083 [3] WANG E T, SANDBERG R, LUO S, et al. Alternative isoform regulation in human tissue transcriptomes[J]. Nature, 2008, 456(7221): 470-476. doi: 10.1038/nature07509 [4] PAN Q, SHAI O, LEE L J, et al. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing[J]. Nat Genet, 2008, 40(12): 1413-1415. doi: 10.1038/ng.259 [5] SCOTTI M M, SWANSON M S. RNA mis-splicing in disease[J]. Nat Rev Genet, 2016, 17(1): 19-32. doi: 10.1038/nrg.2015.3 [6] JUMBE S L, PORAZINSKI S R, OLTEAN S, et al. The evolutionarily conserved cassette exon 7b drives ERG's oncogenic properties[J]. Transl Oncol, 2019, 12(1): 134-142. doi: 10.1016/j.tranon.2018.09.001 [7] YUAN J H, LIU X N, WANG T T, et al. The MBNL3 splicing factor promotes hepatocellular carcinoma by increasing PXN expression through the alternative splicing of lncRNA-PXN-AS1[J]. Nat Cell Biol, 2017, 19(7): 820-832. doi: 10.1038/ncb3538 [8] DAVID C J, CHEN M, ASSANAH M, et al. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer[J]. Nature, 2010, 463(7279): 364-368. doi: 10.1038/nature08697 [9] YANG Q, ZHAO J, ZHANG W, et al. Aberrant alternative splicing in breast cancer[J]. J Mol Cell Biol, 2019, 11(10): 920-929. doi: 10.1093/jmcb/mjz033 [10] TILGNER H, KNOWLES D G, JOHNSON R, et al. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs[J]. Genome Res, 2012, 22(9): 1616-1625. doi: 10.1101/gr.134445.111 [11] SALDI T, CORTAZAR M A, SHERIDAN R M, et al. Coupling of RNA polymerase II transcription elongation with pre-mRNA splicing[J]. J Mol Biol, 2016, 428(12): 2623-2635. doi: 10.1016/j.jmb.2016.04.017 [12] LOERCH S, LEACH J R, HORNER S W, et al. The pre-mRNA splicing and transcription factor Tat-SF1 is a functional partner of the spliceosome SF3b1 subunit via a U2AF homology motif interface[J]. J Biol Chem, 2019, 294(8): 2892-2902. doi: 10.1074/jbc.RA118.006764 [13] ANDERSSON R, ENROTH S, RADA-IGLESIAS A, et al. Nucleosomes are well positioned in exons and carry characteristic histone modifications[J]. Genome Res, 2009, 19(10): 1732-1741. doi: 10.1101/gr.092353.109 [14] SCHOR I E, RASCOVAN N, PELISCH F, et al. Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing[J]. Proc Natl Acad Sci USA, 2009, 106(11): 4325-4330. doi: 10.1073/pnas.0810666106 [15] ZHU H, HINMAN M N, HASMAN R A, et al. Regulation of neuron-specific alternative splicing of neurofibromatosis type 1 pre-mRNA[J]. Mol Cell Biol, 2008, 28(4): 1240-1251. doi: 10.1128/MCB.01509-07 [16] LUCO R F, PAN Q, TOMINAGA K, et al. Regulation of alternative splicing by histone modifications[J]. Science, 2010, 327(5968): 996-1000. doi: 10.1126/science.1184208 [17] CIEŚLIK M, BEKIRANOV S. Combinatorial epigenetic patterns as quantitative predictors of chromatin biology[J]. BMC Genomics, 2014, 15(1): 1-18. doi: 10.1186/1471-2164-15-1 [18] ZHU Y, SUN L, CHEN Z, et al. Predicting enhancer transcription and activity from chromatin modifications[J]. Nucleic Acids Res, 2013, 41(22): 10032-10043. doi: 10.1093/nar/gkt826 [19] SHINDO Y, NOZAKI T, SAITO R, et al. Computational analysis of associations between alternative splicing and histone modifications[J]. FEBS Lett, 2013, 587(5): 516-521. doi: 10.1016/j.febslet.2013.01.032 [20] SAMMETH M, FOISSAC S, GUIGó R. A general definition and nomenclature for alternative splicing events[J]. PLoS Comput Biol, 2008, 4(8): e1000147. doi: 10.1371/journal.pcbi.1000147 [21] CHAUDHARY S, KHOKHAR W, JABRE I, et al. Alternative splicing and protein diversity: Plants versus animals[J]. Front Plant Sci, 2019, 10: 708. doi: 10.3389/fpls.2019.00708 [22] ENROTH S, BORNELöV S, WADELIUS C, et al. Combinations of histone modifications mark exon inclusion levels[J]. PLoS One, 2012, 7(1): e29911. doi: 10.1371/journal.pone.0029911 [23] CHEN W, FENG P, DING H, et al. Classifying included and excluded exons in exon skipping event using histone modifications[J]. Front Genet, 2018, 9: 433. doi: 10.3389/fgene.2018.00433 [24] CHEN W, SONG X, LIN H. Combinatorial pattern of histone modifications in exon skipping event[J]. Front Genet, 2019, 10: 122. doi: 10.3389/fgene.2019.00122 [25] LUCO R F, ALLO M, SCHOR I E, et al. Epigenetics in alternative pre-mRNA splicing[J]. Cell, 2011, 144(1): 16-26. doi: 10.1016/j.cell.2010.11.056 [26] XU Y, ZHAO W, OLSON S D, et al. Alternative splicing links histone modifications to stem cell fate decision[J]. Genome Biol, 2018, 19(1): 133. doi: 10.1186/s13059-018-1512-3 [27] AGIRRE E, OLDFIELD A J, BELLORA N, et al. Splicing-associated chromatin signatures: A combinatorial and position-dependent role for histone marks in splicing definition[J]. Nat Commun, 2021, 12(1): 682. doi: 10.1038/s41467-021-20979-x [28] BOLGER A M, LOHSE M, USADEL B. Trimmomatic: A flexible trimmer for Illumina sequence data[J]. Bioinformatics, 2014, 30(15): 2114-2120. doi: 10.1093/bioinformatics/btu170 [29] WAGNER G P, KIN K, LYNCH V J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples[J]. Theory Biosci, 2012, 131(4): 281-285. doi: 10.1007/s12064-012-0162-3 [30] PATRO R, DUGGAL G, LOVE M I, et al. Salmon provides fast and bias-aware quantification of transcript expression[J]. Nat Methods, 2017, 14(4): 417-419. doi: 10.1038/nmeth.4197 [31] TRINCADO J L, ENTIZNE J C, HYSENAJ G, et al. SUPPA2: Fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions[J]. Genome Biol, 2018, 19(1): 1-11. doi: 10.1186/s13059-017-1381-1 [32] QUINLAN A R, HALL I M. BEDTools: A flexible suite of utilities for comparing genomic features[J]. Bioinformatics, 2010, 26(6): 841-842. doi: 10.1093/bioinformatics/btq033 [33] YU H, ZHU S, ZHOU B, et al. Inferring causal relationships among different histone modifications and gene expression[J]. Genome Res, 2008, 18(8): 1314-1324. doi: 10.1101/gr.073080.107 [34] FRANZ M, LOPES C T, HUCK G, et al. Cytoscape. js: A graph theory library for visualisation and analysis[J]. Bioinformatics, 2016, 32(2): 309-311. [35] GONZALEZ I, MUNITA R, AGIRRE E, et al. A lncRNA regulates alternative splicing via establishment of a splicing-specific chromatin signature[J]. Nat Struct Mol Biol, 2015, 22(5): 370-376. doi: 10.1038/nsmb.3005 [36] KOLASINSKA-ZWIERZ P, DOWN T, LATORRE I, et al. Differential chromatin marking of introns and expressed exons by H3K36me3[J]. Nat Genet, 2009, 41(3): 376-381. doi: 10.1038/ng.322 [37] SIMS R J III, MILLHOUSE S, CHEN C F, et al. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing[J]. Mol Cell, 2007, 28(4): 665-676. doi: 10.1016/j.molcel.2007.11.010 [38] DAVIE J R, XU W, DELCUVE G P. Histone H3K4 trimethylation: Dynamic interplay with pre-mRNA splicing[J]. Biochem Cell Biol, 2016, 94(1): 1-11. doi: 10.1139/bcb-2015-0065 -
 点击查看大图
点击查看大图
图(4) / 表(1)
计量
- 文章访问数: 3923
- HTML全文浏览量: 1092
- PDF下载量: 51
- 被引次数: 0


