 ISSN
ISSN 


-
固体金属表面的氢吸附,属于表面吸附的基础性研究,在催化、防腐、氢储存及运输、核裂变技术、能源电池等方面具有重要的现实意义,因此,关于氢与各种金属表面的相互作用研究一直受到人们的广泛关注[1-5]。对于氢与过渡金属Cu表面的相互作用研究,在实验和理论上都已有很多报道。在实验方面,电子损失谱、振动谱、激光诱导热脱附谱、低能电子衍射等表面技术被应用到氢在Cu表面的吸附研究中[6-8],对氢在Cu表面的吸附结构、表面扩散机理及其H2的解离吸附现象等方面做了详细的研究工作,得到了大量的实验数据。在理论方面,各种理论模拟方法对铜-氢体系也进行了研究。文献[9]用平面波、梯度相关的密度泛函理论计算得出,在Cu (110) 面上,氢原子最稳定的吸附位为短桥位和赝式三重位,但计算得到的振动频率只有赝式三重位和实验符合得较好。文献[10]采用5-MP势方法,对原子氢在金属Cu的3个低指数面上的吸附特性进行了比较系统的研究,结果显示低温低覆盖条件下,氢原子在Cu (110) 表面上只存在赝式三重位和长桥位吸附态,没有短桥位吸附态,并且获得了实验和理论的支持。文献[11]采用准经典轨线法和量子散射理论研究了H2在Cu (110) 的碰撞反应,得到了H2在Cu (110) 表面散射碰撞后的稳定几何构型、能量、谐振频率等,并和实验吻合得较好。
上述研究是在不同条件或者不同方法下进行,所以一些实验和计算的结果是有差别的,甚至有的结论是矛盾的,而且关注的多是氢原子在Cu表面的吸附问题,对H2在Cu表面的研究较少。本文拟重点关注H2在Cu (111) 表面的相互作用,采用基于密度泛函理论的第一性原理来研究H2在Cu (111) 表面的吸附与解离的情况,计算了H2在Cu (111) 表面的临界解离距离。
-
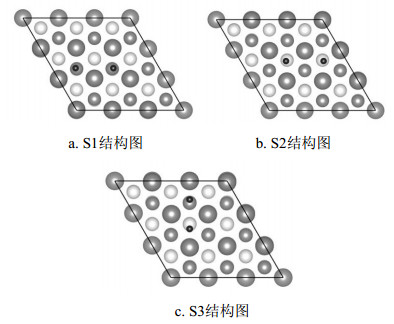
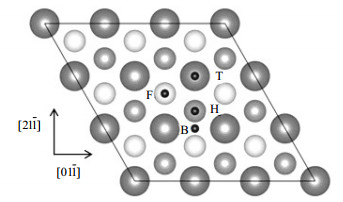
考虑到边界效应和周期性结构问题,采用Cu (111)-(3x3) 超原胞表面模型,由6层Cu原子和15 Å的真空层组成,其中上面3层Cu原子和吸附H2分子可以自由弛豫,下面3层Cu原子固定。Cu (111) 面上的H2分子的初始位置考虑了4种不同的高对称位置[12],如图 1所示,其中T表示顶位 (top),B表示桥位 (bri),F代表面心位 (fcc),H代表六角位 (hcp)。图中还标出了两个平行于Cu (111) 表面的x[011]和y[21 1]方向。

图 1 Cu (111) 表面吸附位置:用黑色小球标识,灰色大球代表Cu (111) 表面顶层Cu原子,中等深色和浅色球分别代表次层和第3层Cu原子。
计算使用基于密度泛函理论的VASP软件包[13](维也纳从头计算程序包)。采用的赝势为VASP软件包赝势库中的标准赝势,电子和核之间的相互作用采用投影缀加平面波方法 (PAW)[14],交换关联泛函为广义梯度近似 (GGA) 的PBE方法[15]。采用选择的赝势得到fcc结构Cu的晶格常数为3.635 Å,这与实验值3.615符合得较好[10];单个H2的键长和解离能分别为0.740 Å和4.526 eV,与实验值0.741 Å和4.546 eV的差别很小[16]。计算中运用了6x6x1布里渊区网格设置和Monkhorst-Pack方案[17]自动产生的不可约k点作自洽计算。平面波展开的截断能设置为400 eV,总能收敛性判据为10-5 eV·atom-1,力的收敛判据为0.01 eV·atom-1。
关于H2在Cu (111) 表面的吸附,其吸附能 (Eads) 采用以下公式计算得到[18]:
$$ {E_{{\rm{ads}}}} = {E_{{\rm{Cu}} + {{\rm{H}}_2}}}-{E_{{\rm{Cu}}}}-{E_{{{\rm{H}}_2}}} $$ (1) 式中,${E_{{\rm{Cu}}}}$和${E_{{{\rm{H}}_2}}}$分别表示Cu (111) 表面的总能和一个自由H2的总能;${E_{{\rm{Cu}} + {{\rm{H}}_2}}}$表示吸附了H2之后的Cu (111) 表面的总能量。
对于一个H2解离为两个氢原子的情况,每个氢原子吸附在Cu (111) 表面的吸附能的计算公式为:
$$ E_{{\rm{ads}}}^{\rm{*}} = ({E_{{\rm{Cu}} + 2{\rm{H}}}}-{E_{{\rm{Cu}}}}-2{E_{\rm{H}}})/2 $$ (2) 式中,${E_{{\rm{Cu}} + 2{\rm{H}}}}$表示H2解离为两个氢原子吸附在Cu (111) 表面的体系总能量;${E_{{\rm{H}}}}$表示一个自由氢原子的能量;${E_{{\rm{Cu}}}}$含义同式 (1)。
-
采用以上方法研究了H2与Cu (111) 表面的相互作用,其中H2的初始构型分为垂直或平行于Cu (111) 表面。为了模拟研究H2在Cu (111) 表面的吸附和解离的情况,根据稳定吸附在Cu (111) 表面的单个氢原子与表面的垂直距离 (大约为0.9~1.5 Å[10]),初始位置的H2中心与表面的距离 (hH) 设为0.9 Å。如果H2不发生分解,就以0.2 Å的间隔不断减小hH,直至H2中最靠近Cu表面的氢原子处在表面之上。对不同初始构型进行弛豫结构优化后,对H2与Cu (111) 表面的相互作用、H2的最终构型进行了详细分析。
-
H2垂直置于Cu (111) 表面的4个高对称吸附位分别标记为bri-z,fcc-z,hcp-z和top-z,初始hH=0.9 Å。表 1列出了驰豫后的H2的终态结构及其参数,包括两个氢原子之间的距离 (dH-H),最近邻的Cu、H原子之间的距离 (dCu-H),H2中心距离Cu (111) 表面的垂直高度 (hH),H2与表面间的吸附能 (Eads) 等参数。
表 1 H2垂直吸附在Cu (111) 表面的末态稳定结构及其参数
初始吸附位置 末态稳定结构 dH-H /Å dCu-H/Å hH /Å Eads/eV bri-z H2 0.753 3.083 3.163 -0.018 fcc-z H2 0.754 3.041 3.024 -0.009 hcp-z H2 0.754 3.111 3.082 -0.012 top-z H2 0.758 2.799 3.167 -0.018 从表 1可以看出,所有情况下,H2驰豫后两原子间的距离dH-H均在0.75~0.76Å之间,略大于自由H2的键长0.74 Å,表示H2没有发生解离,是以分子形式吸附于表面。这是由于此初始距离下表面Cu原子距离H2较远,作用较弱,无法使氢-氢键断裂,因此驰豫后H2结构稳定,这与文献[19]的计算结果是一致的。稳定后表面Cu原子与氢原子的最邻近距离dCu-H大约为3.0 Å,该距离大于H-和Cu2+的离子半径之和 (~2.4 Å),这进一步表明H2和Cu原子之间只存在很弱的相互作用力。H2中心到Cu (111) 表面的距离hH大约在2.8~3.1 Å,模拟过程显示H2是从初始位置整体沿[111]方向向上移动,直到距离表面3.0 Å左右的位置达到平衡稳定下来,形成在Cu (111) 表面的H2吸附,这与H2吸附在Mg (0001) 表面的情况是类似的[20]。四个高对称位H2的吸附能Eads均为负值,说明H2和表面相互作用过程中是放热的[21],并且桥位和顶位的吸附能比其他位置要小很多,表明H2更倾向于吸附在Cu (111) 表面的桥位和顶位。综合上述可以得出,H2在Cu (111) 表面的垂直吸附为相互作用较弱的物理吸附。
为了研究H2在Cu (111) 表面垂直吸附情况下的解离情况,把H2初始吸附位置距离Cu (111) 表面的垂直高度hH从0.9 Å逐渐缩小,结果发现,直到hH减小到0.37 Å(即H2中靠近表面的氢原子已经在Cu表面上) 的过程中,H2都没有发生解离现象,而且对于不同的初始吸附高度,氢分子经过弛豫最终的稳定结构和参数都基本相同。这表明垂直于Cu (111) 吸附的H2不会发生解离现象,只能以分子形式吸附在Cu (111) 表面上。
-
H2平行于Cu (111) 表面吸附考虑了相互垂直的两个方向,即H2的轴线分别沿着Cu (111) 表面的[011](x) 和[21 1](y) 晶向,如图 1所示。H2沿x和y方向平行置于Cu (111) 表面的四个高对称吸附位分别标记为bri-x,fcc-x,hcp-x,top-x和bri-y,fcc-y,hcp-y,top-y,初始hH=0.9 Å。表 2给出了H2沿x方向平行置于Cu (111) 表面,经驰豫后H2的终态结构及其参数,其中hc表示H2解离的临界距离,其他参数含义同表 1。从表中可以看到,H2在fcc、hcp和top位均未解离。H2的键长dH-H及吸附能Eads和H2垂直吸附的时候大小基本相当,但氢原子与最近邻Cu原子的距离dCu-H和H2到表面的高度hH却明显增大了,这说明H2在Cu (111) 表面更倾向于垂直吸附。
表 2 H2沿[011]晶向平行吸附在Cu (111) 表面的末态稳定结构及其参数
初始吸附位置 末态稳定结构 dH-H/Å dCu-H/Å hH /Å Eads/eV hc /Å bri-x H2 0.751 3.396 3.263 -0.010 fcc-x H2 0.750 3.577 3.371 -0.015 0.75 S1 2.707 1.714 0.984 -2.50 hcp-x H2 0.751 3.616 3.398 -0.016 0.75 S2 2.722 1.717 1.002 -2.51 top-x H2 0.751 3.871 3.861 -0.026 0.65 S2 2.716 1.711 0.954 -2.51 为了获得H2沿[011]晶向放置在fcc、hcp及top位的临界解离距离hc,将hH从0.9 Å逐渐减小,得到的临界解离距离和解离后的结构数据如表 2所示。fcc和hcp位的H2解离距离为0.75 Å,top位为0.65 Å,这说明H2由远及近的靠近Cu (111) 表面时,fcc和hcp位置吸附的H2会先解离,然后才是top位,这与fcc和hcp位在H2沿[011]晶向放置时的受力情况有关。
图 2给出了H2解离成两个氢原子后,氢原子弛豫稳定后出现的3种可能结构图S1、S2和S3,分别如图 2a、图 2b、图 2c所示,S1结构代表解离后的两个氢原子吸附在近邻的两个hcp位 (hcp+hcp),S2结构代表解离后的两个氢原子稳定吸附在近邻的两个fcc位 (fcc+fcc),S3结构代表解离后的两个氢原子其中一个吸附在hcp位,另外一个吸附在近邻的fcc位 (hcp+fcc)。

图 2 H2解离后的两个氢原子在Cu (111) 表面稳定吸附的构型图,黑色小球代表氢原子,其他各球代表同图 1
比较表 2中H2的临界解离距离hc和解离后两个氢原子到Cu (111) 表面的平均距离hH(~1.0 Å),发现H2解离后的两个氢原子是垂直表面向上移动达到稳定吸附位。而且fcc位H2解离后的两个氢原子经过弛豫,最后稳定吸附在相邻的两个hcp位,形成hcp+hcp的S1构型,如图 2a,而hcp和top位的H2解离后两个氢原子最后稳定在相邻的两个fcc位,形成fcc+fcc的S2构型,如图 2b。这可能是因为hcp和fcc恰好都是氢原子在Cu (111) 表面的最稳定吸附位置[22]。解离后的两个氢原子之间的距离dH-H超过了2.7 Å,数倍于自由H2的键长,这意味着解离后的两个氢原子之间已经没有相互作用[20]。氢原子与最近邻Cu原子的距离dCu-H为1.7 Å,该距离小于于H-和Cu2+的离子半径之和 (~2.4 Å),说明H和Cu原子之间存在相互作用力,这和分子吸附的情况不同。弛豫稳定后两个氢原子距离Cu (111) 表面的平均距离大约为1 Å,S1和S2中每个氢原子的吸附能达到了-2.5 eV,而且fcc、hcp和top位的吸附能基本是相同的, 解离后氢原子的这些吸附特性与文献[9, 22-23]中的研究结论是相符合的。综合以上数据,可以得出,解离后的氢原子与Cu (111) 表面形成了稳固的化学吸附。然而,在沿x方向平行置于bri位置上,初始hH减小到0.0 Å,H2都没有发生解离,而最终以分子形式物理吸附在Cu (111) 表面,这可能是因为H2沿x方向置于bri位时,该位置的高度对称性使得H2水平方向受到来自表面Cu原子作用力也是高度对称,而且是对H2形成压缩的作用效果,所以H2初始时刻不但不会解离,反而出现了两个氢原子还会相互靠近,同时H2不断向上移动,直到达到稳定位置。
同样的方法对H2沿[21 1]晶向平行置于Cu (111) 表面的情况进行了研究,表 3给出了稳定后H2吸附能及其结构参数。比较表 2,H2沿[21 1]晶向与沿[011]晶向解离吸附稳定后的各种参数存在许多不同,这表示H2在Cu (111) 表面的解离吸附行为与H2的轴向是密切相关的。首先,H2发生解离的位置是bri、fcc和hcp位,没有发生解离的位置是top位,而且解离后的两个氢原子弛豫后的稳定构型为S3(hcp+fcc),如图 2c。其次,[21 1]方向的H2的临界解离距离hc相对[011]方向H2的hc明显增大,fcc和hcp位为0.85 Å,而bri位却达到了1.35 Å,远远大于所有其他位置的H2的临界解离距离,说明H2沿[21 1]晶向平行靠近Cu (111) 表面时,bri位是最容易解离的。文献[24]研究H2在Mg (0001) 面上吸附行为的时候也是首先发现了bri位的解离现象。这是因为此吸附结构恰好使得H2的两个氢原子受到来自近邻Cu原子方向相反的一对作用力,从而使得氢-氢键容易被撕裂断开的缘故。再者,从吸附能来看,氢原子S3构型的吸附能比S1和S2构型大,说明S1和S2吸附构型比S3更稳定。此外,S3中两个氢原子之间的稳定距离dH-H约为2.0 Å,比S1和S2中的dH-H小,这可能是导致S3没有S1和S2稳定的原因。对于H2沿[21 1]晶向平行吸附在Cu (111) 表面,各个参数与沿[011]晶向平行吸附的情况差别较小,这表明H2的轴向对物理吸附的影响不大。
表 3 H2沿[211]晶向平行吸附在Cu (111) 表面的末态稳定结构及其参数
初始吸附位置 末态稳定结构 dH-H/Å dCu-H/Å hH /Å Eads/eV hc /Å bri-y H2 0.751 3.578 3.307 -0.013 1.35 S3 1.995 1.809 0.965 -2.36 fcc-y H2 0.752 3.682 3.488 -0.017 0.85 S3 2.029 1.630 1.014 -2.36 hcp-y H2 0.752 3.322 3.105 -0.009 0.85 S3 2.022 1.635 1.037 -2.35 top-y H2 0.750 3.522 3.510 -0.021 -
采用基于密度泛函理论的第一性原理计算研究了Cu (111) 表面与H2的相互作用,得到了H2在Cu (111) 表面高对称位置的吸附能、稳定吸附结构及其解离情况等性质。计算结果表明H2是否解离取决于H2距表面的初始距离和其初始构型。未解离的氢分子与Cu (111) 表面之间形成作用较弱的物理吸附,而解离后的氢原子与Cu (111) 表面之间形成作用较强的化学吸附。H2物理吸附的吸附能远远小于氢原子化学吸附在Cu (111) 表面的吸附能,而且弛豫稳定后的位置距离表面的高度也明显不同,H2物理吸附高度大约在3.0~4.0 Å,氢原子化学吸附高度大约是1.0 Å。H2垂直于Cu (111) 表面以及沿[011]方向平行放置在bri位和沿[21 1]方向平行放置top位的时候,H2不会发生解离,形成物理吸附。H2平行于Cu (111) 表面放置的时候,H2是否解离主要取决于H2与表面间的初始距离和H2的轴向,H2沿[21 1]方向平行靠近表面时临界解离距离为1.35 Å,其他情况下临界解离距离为0.65~0.85 Å。
First-Principles Study on H2 Adsorption and Dissociation on Cu (111) Surface
-
摘要: 采用基于密度泛函理论的第一性原理对氢分子与Cu (111) 表面的相互作用进行了研究。计算结果表明氢分子是否解离取决于氢分子距表面的初始距离和其初始构型。当氢分子垂直于Cu (111) 表面放置时,在距离表面0.37~4.0 Å范围内,氢分子与Cu (111) 表面相互作用后均不会解离,物理吸附在Cu (111) 表面;当氢分子平行于Cu (111) 表面放置时,有的氢分子解离成氢原子后化学吸附于表面六角 (hcp) 或面心 (fcc) 位。氢分子在桥位 (bri) 并沿[211]方向平行靠近Cu (111) 表面时,氢分子解离的临界距离约为1.35 Å,其他情况下在0.65~0.85 Å之间。
-
关键词:
- 吸附 /
- Cu (111) 表面 /
- 解离 /
- 第一性原理 /
- 氢分子
Abstract: The interactions of hydrogen molecules (H2) and the Cu (111) surface have been studied by using density functional theory based first-principles calculations. The results show that the dissociation of H2 depends mainly on the initial distance from H2 to the surface (hH) and the initial H2 configurations. The H2 molecules of vertical adsorption on Cu (111) are not dissociated for the initial hH of 0.3-4.0 Å, and the undissociated H2 is physically adsorbed on the Cu (111) surface. When H2 molecules are adsorbed on Cu (111) surface in parallel, some of the H2 are dissociated into two hydrogen atoms, which occupy hcp and fcc sites and form stable chemisorption on the Cu (111) surface. The critical distance of H2 dissociation at bri sites is 1.35 Å for parallel adsorption along the[211] direction and 0.65-0.86 Å for other cases.-
Key words:
- adsorption /
- Cu (111) surface /
- dissociation /
- first-principles /
- hydrogen molecule
-
表 1 H2垂直吸附在Cu (111) 表面的末态稳定结构及其参数
初始吸附位置 末态稳定结构 dH-H /Å dCu-H/Å hH /Å Eads/eV bri-z H2 0.753 3.083 3.163 -0.018 fcc-z H2 0.754 3.041 3.024 -0.009 hcp-z H2 0.754 3.111 3.082 -0.012 top-z H2 0.758 2.799 3.167 -0.018  下载: 导出CSV
下载: 导出CSV
表 2 H2沿[011]晶向平行吸附在Cu (111) 表面的末态稳定结构及其参数
初始吸附位置 末态稳定结构 dH-H/Å dCu-H/Å hH /Å Eads/eV hc /Å bri-x H2 0.751 3.396 3.263 -0.010 fcc-x H2 0.750 3.577 3.371 -0.015 0.75 S1 2.707 1.714 0.984 -2.50 hcp-x H2 0.751 3.616 3.398 -0.016 0.75 S2 2.722 1.717 1.002 -2.51 top-x H2 0.751 3.871 3.861 -0.026 0.65 S2 2.716 1.711 0.954 -2.51
下载: 导出CSV
表 3 H2沿[211]晶向平行吸附在Cu (111) 表面的末态稳定结构及其参数
初始吸附位置 末态稳定结构 dH-H/Å dCu-H/Å hH /Å Eads/eV hc /Å bri-y H2 0.751 3.578 3.307 -0.013 1.35 S3 1.995 1.809 0.965 -2.36 fcc-y H2 0.752 3.682 3.488 -0.017 0.85 S3 2.029 1.630 1.014 -2.36 hcp-y H2 0.752 3.322 3.105 -0.009 0.85 S3 2.022 1.635 1.037 -2.35 top-y H2 0.750 3.522 3.510 -0.021
下载: 导出CSV
-
[1] KRESSE G, HAFNER J. First-principles study of the adsorption of atomic H on Ni (111), (100) and (110)[J]. Surf Sci, 2000, 459(3):287-302. doi: 10.1016/S0039-6028(00)00457-X [2] LI Xiang-gui, ZHANG Ping, CHAN C K. First-principles calculation of Mg (0001) thin films:Quantum size effect and adsorption of atomic hydrogen[J]. Physica B, 2007, 390(1-2):225-230. doi: 10.1016/j.physb.2006.08.019 [3] HUDA M N, RAY A K. A density functional study of atomic hydrogen adsorption on plutonium layers[J]. Physica B, 2004, 352(1-4):5-17. doi: 10.1016/j.physb.2004.06.010 [4] YANG Li, PENG Shu-ming, LONG Xing-gui, et al. Ab initio study of intrinsic, H, and He point defects in hcp-Er[J]. J Appl Phys, 2010, 107(5):054903. doi: 10.1063/1.3309834 [5] LUO Jian, ZHOU Hong-bo, LIU Yue-lin, et al. Dissolution, diffusion and permeation behavior of hydrogen in vanadium:a first-principles investigation[J]. J Phys:Condens Matter, 2011, 23(13):135501. doi: 10.1088/0953-8984/23/13/135501 [6] ASTALDI C, BIANCO A, MODESTI S, et al. Vibrational-spectra of atomic H and D on Cu (110)-evidence for H quantum delocalization[J]. Phys Rev Lett, 1992, 68(1):90-93. doi: 10.1103/PhysRevLett.68.90 [7] JOHNSON D F, CARTER E A. Hydrogen in tungsten:Absorption, diffusion, vacancy trapping, and decohesion[J]. J Mater Res, 2010, 25(2):315-327. doi: 10.1557/JMR.2010.0036 [8] LLOYD P B, SWAMINATHAN M, KRESS J W, et al. Temperature programmed desorption study of the adsorption and absorption of hydrogen on and in Cu (111)[J]. Applied Surface Science, 1997, 119:267-275. doi: 10.1016/S0169-4332(97)00178-5 [9] BAE C S, FREEMAN D L, DOLL J D, et al. Energetics of hydrogen chemisorbed on Cu (110):a first principles calculations study[J]. J Chem Phys, 2000, 113(16):6926-6932. doi: 10.1063/1.1311293 [10] 王泽新, 张晓明, 庞雪辉.原子H在Cu (100) (111) (110) 上的吸附扩散研究[J].化学学报, 2004, 26(4):436-441. http://www.cnki.com.cn/Article/CJFDTOTAL-HXXB200404018.htm WANG Ze-xin, ZHANG Xiao-ming, PANG Xue-hui. Adsorption and diffusion studies of atomic hydrogen on Cu (100) (111) (110)[J]. Acta Optica Sinica, 2004, 26(4):436-441. http://www.cnki.com.cn/Article/CJFDTOTAL-HXXB200404018.htm [11] KROES G J, PIJPER E. Dissociative chemisorption of H2 on the Cu (110) surface:a quantum and quasiclassical dynamical study[J]. The Journal of Chemical Physics, 2007, 127:164722. doi: 10.1063/1.2798112 [12] LIU Tian-hui, FU Bi-na, ZHANG Dong-hui. Validity of the site-averaging approximation for modeling the dissociative chemisorption of H2 on Cu (111) surface:a quantum dynamics study on two potential energy surface[J]. The Journal of Chemical Physics, 2014, 141:194302. doi: 10.1063/1.4901894 [13] KRESSE G, FURTHERMTILLER J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set[J]. Comput Mater Sci, 1996, 6(1):15-50. doi: 10.1016/0927-0256(96)00008-0 [14] KRESSE G, JOUBERT D. From ultrasoft pseudopotentials to the projector augmented-wave method[J]. Phys Rev B, 1999, 59(3):1758-1775. doi: 10.1103/PhysRevB.59.1758 [15] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J]. Phys Rev Lett, 1996, 77(18):3865. doi: 10.1103/PhysRevLett.77.3865 [16] HUBER K P, HERZBERG G. Molecular structure and molecular spectra Ⅳ:Constants of diatomic molecules[M]. New York:Van Rostrand-Reinhold, 1979. [17] MONKHORST H J, Pack J D. Special points for Brillouinzone integrations[J]. Phys Rev B, 1976, 13(12):5188-5192. doi: 10.1103/PhysRevB.13.5188 [18] WANG Sheng-guang, CAO Dong-bo, LI Yong-wang, et al. Chemisorption of CO2 on Nickel surfaces[J]. J Phys Chem B, 2005, 109(40):18956-18963. doi: 10.1021/jp052355g [19] ZUO Zhi-jun, HUANG Wei, HAN Pei-de. A DFT study the solvent effects of H-2 adsorption on Cu (h k l) surface[J]. Appl Surf Sci, 2012, 258(8):3364-3367. doi: 10.1016/j.apsusc.2011.09.024 [20] ZHANG Jian, ZHOU Dian-wu, HUANG Ya-ni, et al. Study on H2 adsorption and dissociation properties on Mg (0001) surface[J]. Rare Metal Mat Eng, 2009, 38(9):1518-1525. doi: 10.1016/S1875-5372(10)60049-5 [21] COTTON F A, WILKINSON G. Advanced inorganic chemistry[M]. New York:Wiley, 1988. [22] GUNDERSEN K, HAMMER B, JACOBSEN K W. Chemisorption and vibration of hydrogen on Cu (111)[J]. Surf Sci, 1993, 285(1-2):27-30. doi: 10.1016/0039-6028(93)90910-C [23] NIE Jin-lan, XIAO Hai-yan, ZU Xiao-tao. First-principles study of H adsorption on and absorption in Cu (111) surface[J]. Chemical Physics, 2006, 321:48-54. doi: 10.1016/j.chemphys.2005.07.027 [24] DU A J, SMITH S C, YAO X D, et al. The role of Ti as a catalyst for the dissociation of hydrogen on a Mg (0001) surface[J]. Journal Physics Chemical B, 2005, 109(38):18037-18041. doi: 10.1021/jp052804c -
 点击查看大图
点击查看大图
图(2) / 表(3)
计量
- 文章访问数: 5389
- HTML全文浏览量: 3749
- PDF下载量: 361
- 被引次数: 0
