 ISSN
ISSN 


 下载:
下载:
-
生物学研究的实用价值之一是应用于医学研究,造福人类健康。识别与疾病相关的基因,是复杂疾病病理学研究中的重要任务之一,它是进行疾病预防、临床治疗和药物设计的前期工程[1]。疾病基因预测实质上是一个优选问题,即在众多潜在基因中优选出最有可能与疾病关联的基因。经过科学家长期的努力,目前已获得大量人类疾病的分子基础方面的知识,例如遗传学方面的连锁分析(linkage analysis)研究已识别了许多与疾病相关的染色体区域,有些染色体区域中的疾病基因已得到确认,但仍有许多区域上具体的致病基因是未知的[2-3]。这些染色体区域包含多达数百个基因,要用实验手段去确认其上具体的致病基因,需要耗费大量的人力、物力及时间。因此,采用计算方法预测区域内的疾病基因,使得生物学家可以有选择地进行实验验证,就是很好的方法。
大量研究证实,相同或相关疾病的致病基因,在功能上通常是相似或相关的[4-7]。这种相似或相关可能是物理意义上的直接结合或属于同一蛋白复合物,也可能是存在非直接的相互作用,例如参与相同的代谢通路或细胞过程,可以从多种视角进行量化研究[8-10]。例如,利用蛋白质组学信息探究基因间的相互作用[11]、利用基因表达数据衡量基因间共表达的程度[12-14]、从基因本体注释即GO中挖掘基因间GO term的相似性[15-16]等,这些方法都可以识别功能上相似或相关的基因。尤其是,功能相似或相关的基因在分子网络中的位置往往是相邻或相近的,这使得开发基于分子网络的疾病基因预测算法,成为近年来的热点课题。这类方法主要基于“连接有罪”原则(guilt-by-association)[17-18],即在分子网络的拓扑结构中,寻找与已知疾病基因相邻、相近、或相似的基因,将其预测为疾病基因。
本文将从数据资源、计算方法、验证方法等方面,综述基于分子网络的疾病基因预测所取得的进展,讨论存在的问题及今后发展的方向。
-
本节介绍基于网络的疾病基因预测算法所需要的基础数据,包括背景网络、已知疾病基因和疾病表型相似性数据。
-
在分子网络中进行疾病基因预测,首先需要一个人类全基因组的蛋白-蛋白相互作用(PPI)网络或基因关联网络作为背景网络。目前,通过高通量生物学实验[19]、低通量生物学实验、文献挖掘等多种方法已建立人类的多个PPI数据库,如HPRD[20]、BioGrid[21]、BIND[22]、MINT[23]、IntAct[24]等。然而,现有的数据只是实际存在数据的冰山一角,覆盖率太低,据估计,经实验证实的人类蛋白质相互作用数据只占实际存在的相互作用数据量的0.3%[25];而且,高通量实验通常产生大量的假阳性和假阴性的数据,造成大量的数据噪声。为了解决现有数据覆盖率低、准确性差的问题,一些研究用计算方法融合不同来源的生物学数据,推断基因之间的关联关系,这里的关联关系既包括基因编码的蛋白间物理上的相互作用、也包括它们功能上的相关性,并对其中每一对关联关系赋予置信分,从而构建了更大的加权基因关联网络,如FLN[26]、String[27]、Humannet[28]、Fun-coup[29]、Hippie[30]等。这些无权的PPI网络和加权的基因关联网络已被应用于不同的研究中,作为疾病基因预测的背景网络。
-
已知的疾病基因作为疾病基因预测的先验信息组成种子集,一般由5~30个基因组成。种子数目太少将导致信息量不足以预测出潜在的疾病基因,如果太多会导致网络预测的生物信息异构化而无法正确反应实际的疾病信息[31]。
随着生物信息技术的不断发展,在科学家的努力下集成了多个疾病基因数据库,如人类孟德尔遗传在线数据库[32](online mendelian inheritance in man, OMIM)、遗传关联数据库[33](genetic association database, GAD)、癌基因组解剖项目[34](cancer genome anatomy project, CGAP)、癌症基因谱数据库[35](cancer gene census, CGC)、DisGeNET[36]等。这些数据库中的信息,可以作为疾病基因预测的先验信息,也可以用于构造训练集评估算法优劣。
-
研究表明,引起相同或相似疾病的基因在功能上相似且在染色体上彼此临近,因而疾病之间的表型相似性会导致功能相关的基因在网络上产生模块化结构,形成由疾病相关基因构成的疾病子图[37]。因此,疾病之间的表型相似性信息将有助于疾病基因的预测,尤其对于一些缺少已知致病基因信息的疾病,可用该疾病的相似表型及其致病基因作为信息补充。
文献[38]基于文本挖掘的方法率先总结疾病表型的相似性,他们采用医学主题词表(MeSH)对每一表型的临床特征或性状表现进行描述,形成描述疾病的特征向量,再对表型间的特征向量求余弦值以量化表型间的相似性,建立了5 080个不同疾病表型间相似性的数据库,该数据库可以从Minminer[39]网页中在线下载。
-
本节将介绍仅利用已知疾病基因信息,在背景网络中进行疾病基因预测的算法。即这里的算法不考虑疾病相似性等其他信息。这类算法将疾病候选基因置于背景网络中,根据候选基因与已知疾病基因在网络位置上的拓扑关系,来预测候选基因是疾病基因的可能性。这种对候选基因进行打分的机制模拟热量传播过程,将已知疾病基因看作初始热源,热量通过网络中的边进行传播,节点获得的热量越多,则越有可能与疾病相关。
-
局部预测算法只运用局部的网络拓扑结构信息,筛选与已知致病基因距离最近、最相关的候选基因。
-
基于“连接有罪”的生物学假设,直接邻居法认为与已知疾病基因在背景网络上直接相连的基因,最有可能是潜在的疾病基因。一个候选基因与疾病的亲疏关系,由其与已知疾病基因间的连边总数或者边权和决定,该数值越大则越有可能预测为致病基因。
文献[40]利用直接邻居法对OMIM中289种至少包含两个以上致病基因的疾病进行检验,这些疾病共有1 003个不同的疾病基因。他们分别采用5个不同的PPI数据集作背景网络,对算法进行验证,发现尽管不同数据集在预测准确性上存在差异,但其预测表现均优于随机选择。
文献[26]利用朴素贝叶斯分类器融合了16组不同的基因功能相关数据,构建了一个具有21 657个基因、22 388 609条边的加权基因关联网络。他们以这个足够稠密的网络为背景网络,利用直接邻居算法预测潜在的致病基因,即取候选基因与疾病的关联得分为其与该疾病所有已知致病基因连边的边权和,取得了很好的预测效果。
-
基因间没有直接的相互作用但是参与同一生物学过程,例如属于同一信号通路或代谢通路的基因,也可能功能相关,它们的功能相关性可以由其最短路径衡量。文献[41]最早用最短路径方法对阿尔茨海默氏病的疾病基因进行预测。以该疾病已知的四个疾病基因(APP, APOE, PSEN1, PSEN2) 作为种子集,首先给每个种子基因赋予一个初始证据分,代表它们与该疾病的关联强度。其次,结合距离衰减函数$ f $计算候选基因与所有种子基因的初始证据分之和,表达为:
$$ E(g)=\sum\limits_{v\in G({{d}_{c}})}^{{}}{{{E}_{{{d}_{c}}}}(v)f({{d}_{gv}})} $$ (1) 式中,$ {d_c} $代表要研究的疾病;$ G({d_c}) $代表由已知疾病基因组成的种子集;$ g $是候选基因;$ {E_{{d_c}}}(v) $是种子节点$ v $的初始证据分;$ {d_{gv}} $是$ g $与$ v $之间的最短距离;$ f({d_{gv}}) = 1/({d_{gv}} + 1) $,是距离衰减函数。它对距离较大的节点进行惩罚和抑制。文献[41]考虑了多种类型的衰减函数如sigmoid函数和线性函数,通过对比发现各种衰减函数所得到的结果差别不大,对$ f $的具体形式不敏感,因此采用这个无参数的距离衰减函数已经可以满足要求。该方法模拟信息从种子集向候选基因沿着最短路径扩散,成功地筛选出那些虽然不是种子节点的直接邻居,但是与种子节点关联程度充分大的那些节点。
-
相比局部预测方法,全局预测算法在全局范围内运用网络的拓扑结构分析候选基因与已知疾病基因的亲疏关系。全局方法能够扩大候选基因的范围以免遗漏那些连接度较低、位于网络边缘的节点,提高准确性[42]。运用较广的全局方法有扩散核算法(diffusion kernel, DK)[43]、重启的随机游走(random walk with restart, RWR)[43]、网络传播算法(network propagation, NP)[44]、Katz指标[45]等。
-
网络的扩散核矩阵,是用懒惰的随机游走(lazy random walk)[46]度量节点对在网络中的邻近程度,其定义为:
$$ \mathit{\boldsymbol{K}}={{\rm{e}}^{-\alpha \boldsymbol{L}}} $$ (2) 式中,$ \mathit{\boldsymbol{L}} $为背景网络的拉普拉斯矩阵,定义为$ \mathit{\boldsymbol{L}} = \mathit{\boldsymbol{D}} - \mathit{\boldsymbol{W}} $,$ \mathit{\boldsymbol{D}} $为网络的度矩阵,$ \mathit{\boldsymbol{W}} $为网络的邻接矩阵;$ \alpha $为扩散常量,它决定了扩散速度。
一个懒惰的随机漫步者在节点i以固定的概率$ \beta $($ \beta \le 1/\mathop {\max }\limits_i \{ {d_i}\} $,$ {d_i} $为节点i的连接度)随机地到达其某个邻居节点,而以概率$ 1 - \beta {d_i} $留在节点i,这个随机过程的转移概率矩阵为$ \mathit{\boldsymbol{I}} + \beta \mathit{\boldsymbol{H}} $($ \mathit{\boldsymbol{H}} = - \mathit{\boldsymbol{L}} $)。懒惰的随机游走中,概率$ \beta $随游走的步数衰减,即在第n步游走时,$ \beta = \frac{\alpha }{n} $,则当$ n \to \infty $时,转移概率矩阵收敛到式(2),即有:
$$ \mathop {\lim }\limits_{n \to \infty } \, {{\left( \mathit{\boldsymbol{I}}+\frac{\alpha }{n}\mathit{\boldsymbol{H}} \right)}^{n}}={{\rm{e}}^{\alpha \boldsymbol{H}}}={{\rm{e}}^{-\alpha \boldsymbol{L}}} $$ (3) 因此扩散核矩阵K的(i, j)元素代表懒惰的随机漫步者从节点i游走到节点j的概率,也称为这两个节点间的扩散核距离。
用扩散核矩阵预测疾病基因时,候选基因与疾病的关联得分定义为其与所有疾病基因的核扩散距离总和。由此可见,扩散核方法实质上是在网络的扩散核矩阵上使用的直接邻居算法[47]。文献[43]将核扩散算法推广至疾病基因预测领域,实验结果表明该算法在预测复杂疾病时效果显著优于直接邻居法和最短路径法。
-
RWR算法模拟一个漫步者从初始节点出发, 随机地选择一条边到达其某个邻居节点的过程。在任意时刻,漫步者可以选择以概率$ r $回到初始节点,或者以与网络边权成正比的概率沿着边到达任意一个邻居节点。节点的序列是有限状态的马尔可夫链,具有无记忆性,即下一个状态的概率只由当前节点的状态决定,与之前状态无关。由于在非二部、无向、连通的网络上的随机游走一定可以达到稳态,因此漫步者在网络中游走足够长的时间,其到达每个节点的概率将会收敛到稳态,此稳态的概率向量便可衡量初始节点与其余节点的网络临近性或相似性[48]。
文献[43]将RWR算法成功用于疾病基因预测,初始节点向量为由已知疾病基因组成的种子集,在第t+1步时,网络节点的概率向量为:
$$ {{\mathit{\boldsymbol{x}}}^{t+1}}=(1-r){{\mathit{\boldsymbol{P}}}_{\rm{RW}}}{{\mathit{\boldsymbol{x}}}^{t}}+r{{\mathit{\boldsymbol{x}}}^{0}} $$ (4) 式中,$ {\mathit{\boldsymbol{P}}_{{\rm{RW}}}}(u, v) = w(uv)/W(u) $,表示对背景网络邻接矩阵$ \mathit{\boldsymbol{W}} $进行列和归一化后的转移概率矩阵;$ W(u) $为节点$ u $与所有连通节点的强度之和;$ w(uv) $为$ u $、$ v $连边的权重;$ {\mathit{\boldsymbol{x}}^0} $为种子节点强度的初始向量,若有$ m $个种子节点,则每个种子节点对应的分量为$ 1/m $,其他节点对应的分量为0;$ r $为重启概率。实际计算中,达到稳态即收敛的方式是不断地迭代式(4),直到$ \left| {{\mathit{\boldsymbol{x}}^{t + 1}}-{\mathit{\boldsymbol{x}}^t}} \right| < \alpha $,$ \alpha $是事先确定的一个接近于0的正数。
RWR算法是Google搜索引擎的核心算法PageRank的扩展应用[49-50]。作为从全局衡量节点间相似性的指标,RWR在链路预测领域也发挥着重要作用[51-52]。
-
文献[44]将网络传播算法用于疾病基因预测。该算法与RWR算法相似,它模拟信息在网络中的传播过程。信息从初始节点沿着网络上的边开始传播,在每一时刻,节点不仅向邻居节点传播信息也收到来自其他邻居节点的信息。当信息流达到稳态时,各个节点所获得的信息量便是其与初始节点的临近性或相似性。其具体公式如下:
$$ {{\mathit{\boldsymbol{x}}}^{t+1}}=(1-r){{\mathit{\boldsymbol{P}}}_{\rm{NP}}}{{\mathit{\boldsymbol{x}}}^{t}}+r{{\mathit{\boldsymbol{x}}}^{0}} $$ (5) 式中,$ {\mathit{\boldsymbol{P}}_{{\rm{NP}}}}(u, v) = w(uv)/\sqrt {W(u)W(v)} $,其余符号的含义及循环结束的条件与与RWR方法相同。
-
Katz指标作为基于路径的相似性指标,考虑了节点之间所有路径数并对较短的路径赋予更大的权重,从全局预测节点之间产生连边的可能性。Katz指标充分地考虑了网络的拓扑结构特征,在链路预测领域取得了相当的效果[53-54]。其数学定义为:
$$ \begin{matrix} \mathit{\boldsymbol{A}}={{(\mathit{\boldsymbol{I}}-\varphi \mathit{\boldsymbol{W}})}^{-1}}-\mathit{\boldsymbol{I}}= \\ \varphi \mathit{\boldsymbol{W}}+{{\varphi }^{2}}{{\mathit{\boldsymbol{W}}}^{2}}+{{\varphi }^{3}}{{\mathit{\boldsymbol{W}}}^{3}}+\cdots +{{\varphi }^{n}}{{\mathit{\boldsymbol{W}}}^{n}} \\ \end{matrix} $$ (6) 式中,$ \mathit{\boldsymbol{W}} $是网络的邻接矩阵;$ \varphi $是对于不同长度的路径赋予的权重衰减因子。为了保证数列收敛,$ \varphi $的取值应当小于$ \mathit{\boldsymbol{W}} $的最大特征值的倒数。受Katz指标在社会网络的运用启发,当前越来越多学者将Katz指标引入疾病基因预测领域,如文献[55]利用Katz指标,结合基因表达数据,在蛋白质相互作用网络中进行疾病基因预测;文献[56]将Katz算法扩展至一个融合了疾病表型相似性、疾病基因信息和PPI网络的异构网络中,进行疾病基因预测。
-
大多数基于网络的疾病基因预测算法偏向于网络中心节点,连接度大的节点更容易被筛选为致病基因,而忽视了那些连接度较低的真正的疾病基因。为了减弱这种偏向性,抑制高连接度节点的虚假得分,文献[57]提出3种数据调整策略对候选基因的原始网络得分进行调整。
策略1保持种子节点的度分布,计算原始得分的z-score:
$$ {{\alpha }_{SD}}(g, {{d}_{c}})=\left( \frac{\alpha (g, {{d}_{c}})-{{\mu }_{S}}}{{{\sigma }_{S}}} \right) $$ (7) 在保留种子节点度分布的前提下,随机产生1 000组伪种子节点。$ {\mu _S} $和$ {\sigma _S} $分别为候选基因$ g $根据这1 000组新的种子节点在网络打分中所得的平均分和标准差;$ \alpha (g, {d_c}) $为候选基因$ g $基于原始种子节点在重启的随机游走算法下的得分;$ {\alpha _{{\rm{SD}}}}(g, {d_c}) $是调整种子节点度偏差后$ g $的z-score。
类似地,策略2保持候选基因的度分布,计算原始得分的z-score。对每个候选基因随机产生1 000组与其度分布一致的对照组,并计算对照组网络得分的平均分和标准差,最后得到候选基因网络得分的z-score,作为调整后的得分。
策略3基于特征向量中心性将候选基因$ g $的重启的随机游走得分和不重启的随机游走得分取对数比即$ \log \frac{{{\alpha ^{(r > 0)}}(g, {d_c})}}{{{\alpha ^{(r = 0)}}(g, {d_c})}} $,这一做法的目的是消除由网络中心性引起的对大度节点的偏向。
实验结果表明,总体上3种调整策略的表现相当且均优于重启的随机游走算法,但是会抑制高连接度基因的表现。因此文献[57]进一步提出3种组合策略,对数据调整后的排名和调整之前的排名进行组合优选。组合的中心思想是对连接度较低的基因采取数据调整后的排名,对大度节点采用调整前的排名。这3种组合策略分别基于候选基因的度分布、乐观的优选策略以及基于已知致病基因度分布。验证结果表明基于特征向量中心性进行数据调整并且采取基于已知致病基因度分布的组合策略表现最好,其表现显著优于RWR[43]和NP[44]。
-
表型相似性与基因相似性之间存在一定程度的相关性[18, 58-59]。在疾病基因预测中结合疾病的表型相似性,将有助于增强潜在疾病基因与预测疾病的关联,使得预测更为精准。这类研究中,疾病表型间的相似性信息主要来自文献[38]建立的表型相似分数据库,而疾病的表型相似性信息主要通过两种方式运用到疾病基因预测中。一种方式仍然以PPI网络或基因关联网络为背景网络,直接将表型相似性信息结合到预测算法中。第二种方式是构建一个包含基因-基因、基因-疾病、疾病-疾病3类关系的异构网络作为背景网络,在此网络上分析候选基因与疾病的关联关系。下文将详细介绍基于这两种方式的疾病基因预测方法。
-
这类方法中,种子集通常不仅包含已知的疾病基因,还包含与该疾病相似的其他疾病表型的疾病基因,这对于一些具有较少先验信息的疾病具有重要意义。
-
文献[60]提出的VAVIEN算法利用候选基因与致病基因在网络拓扑上的结构相似性,来衡量候选基因与疾病的关联性。详细的预测步骤如下:
1) 定义基因$ g $与所研究的疾病$ {d_c} $之间的关联分$ \sigma (g, {d_c}) $定义为:
$$ \sigma (g, {{d}_{c}})=\left\{ \begin{align} &1 \\ &\max S({{d}_{c}}, {{d}_{k}})\ \ k=1, 2, \cdots, n\ \\ &0 \\ \end{align} \right. $$ (8) 式中,$ S({d_c}, {d_i}) $为来自Mimminer的疾病表型$ {d_c} $与$ {d_i} $之间的相似性分。即若$ g $为$ {d_c} $的致病基因,则$ g $与$ {d_c} $的关联得分赋值为1;若$ g $是$ {d_c} $的$ n $个相似表型的致病基因,则赋予其中最大的表型相似性得分;否则,赋分为0。
2) 利用RWR算法对网络中每个基因$ g $建立其拓扑结构向量$ {\mathit{\boldsymbol{\beta }}_g} $。
基因$ g $的拓扑结构向量等于随机漫步者从该点出发游走整个网络得到的稳态结果,即节点$ g $到网络中其他节点的概率。
3) 定义网络中任意两基因$ u $和$ v $的拓扑结构相似性$ \rho (u, v) $为它们的拓扑结构向量间的皮尔逊相关系数,即:
$$ \rho (u, v) = {\rm{corr}}({\mathit{\boldsymbol{\beta }}_u}, {\mathit{\boldsymbol{\beta }}_v}) $$ (9) 4) 对每个候选基因$ g $,计算它与疾病$ {d_c} $的种子集$ G({d_c}) $中致病基因平均拓扑结构向量之间的相似性得分:
$$ \alpha (g, {d_c}) = \rho ({\mathit{\boldsymbol{\beta }}_\mathit{\boldsymbol{g}}}, {\mathit{\boldsymbol{\overline \beta }} _{\mathit{\boldsymbol{G}}({d_c})}}) $$ (10) $$ {\mathit{\boldsymbol{\overline \beta }} _{\mathit{\boldsymbol{G}}({d_c})}} = \frac{{\sum\limits_{v \in G({d_c})} {\sigma (v, {d_c}){\mathit{\boldsymbol{\beta }}_v}} }}{{\sum\limits_{v \in G({d_c})} {\sigma (v, {d_c})} }} $$ (11) 式中,$ v $是致病基因;$ {\mathit{\boldsymbol{\overline \beta }} _{\boldsymbol{G}({d_c})}} $代表种子节点的平均拓扑结构向量;$ \alpha (g, {d_c}) $是候选基因$ g $与疾病$ {d_c} $的相似性得分,得分越高,关联越大。文献[60]基于候选基因与种子节点的拓扑结构相似性提出了ATS、TSA、TSR这3种优选基因策略。式(10) 代表TSA,是其中表现最好的一种。实验结果表明VAVIEN的算法表现优于RWR[43],PRINCE(PRIoritization and complex elucidation, PRINCE)[44]和DADA[57]。
-
文献[44]提出的PRINCE算法融合疾病相似性信息于网络传播算法中。与式(5) 的网络传播算法相比,PRINCE算法仅仅是初始向量$ {\mathit{\boldsymbol{x}}^0} $不同。这里的种子集包含已知的疾病基因以及与该疾病相似的其他疾病表型的疾病基因,因此初始向量的定义有变化。
PRINCE采用的表型数据仍然来源于Minminer数据库。van Driel对不同数值的表型相似性的预测能力进行了测试,发现当相似值在[0, 0.3]时信息量不足,而当值落在[0.6, 1]时表型间具有显著的功能相似性。因此,PRINCE算法在融合表型相似性信息时,用Logistic函数抑制相似性值较低的表型、保留具有显著性的表型:
$$ L(x) = \frac{1}{{1 + {{\rm{e}}^{(cx + d)}}}} $$ (12) 式中,$ c $和$ d $为参数,式(12) 使得当$ x \in [0, 0.3] $时,$ L(x) \approx 0 $;当$ x \in [0.6, 1] $时,$ L(x) \approx 1 $。
初始向量$ {\mathit{\boldsymbol{x}}^0} $定义为:
$$ {\boldsymbol{x}^0} = \left\{ \begin{array}{l} 1\\ \max [L(S({d_c}, {d_k}))]\;{\rm{ }}\ \ k = 1, 2, \cdots, n\;\\ 0 \end{array} \right. $$ (13) 假设疾病$ {d_c} $有$ n $个相似表型,$ S({d_c}, {d_i}) $为来自Mimmine的疾病表型$ {d_c} $与$ {d_i} $之间的相似性分,$ L(S({d_c}, {d_i})) $代表经Logistic函数处理后的表型相似性分。若基因$ g $是疾病$ {d_c} $的疾病基因,则$ {\mathit{\boldsymbol{x}}^0} $对应的分量值为1;若$ g $是$ {d_c} $的多个相似表型的致病基因,则$ {\mathit{\boldsymbol{x}}^0} $对应分量取经Logistic函数处理后的表型相似分的最大值;否则为0。通过留一交叉验证,表明PRINCE的预测结果要比RWR[43]和CIPHER[61]效果好。
文献[62]后续提出的ProSim算法是对PRINCE的进一步改进。ProSim在初始向量中不仅考虑了表型的相似性信息,也考虑了所有候选基因与已知致病基因在网络中的邻近性。
-
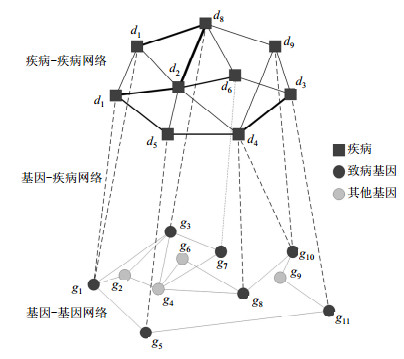
此类方法将PPI网络(或基因关联网络)、疾病与基因关联的二部网络、以及疾病表型相似性网络整合在一起,构建一个包含基因-基因、基因-疾病、疾病-疾病三类关系的异构网络(见图 1),以此网络作为背景网络,分析候选基因与疾病的关联关系。

图 1 异构网络
-
RWRH(random walk with restart on heterogeneous network)是在异构的网络中运用RWR算法进行全局预测,即将RWR的计算式(4) 修改为:
$$ {\mathit{\boldsymbol{p}}_{s + 1}} = (1-r){\mathit{\boldsymbol{M}}^{\rm{T}}}{\mathit{\boldsymbol{p}}_\boldsymbol{S}} + r{\mathit{\boldsymbol{p}}_0} $$ (14) 式中,代表异构网络的转移概率矩阵,$ {\mathit{\boldsymbol{M}}_G} $和$ {\mathit{\boldsymbol{M}}_P} $分别是异构网络中基因-基因和疾病-疾病子网络的转移概率矩阵,$ {\mathit{\boldsymbol{M}}_{GP}} $和$ {\mathit{\boldsymbol{M}}_{PG}} $分别是基因-疾病和疾病-基因二部子网络的的转移概率矩阵;$ {\mathit{\boldsymbol{p}}_0} $代表的是异构网络的初始向量,定义为:。其中$ {\mathit{\boldsymbol{u}}_{\rm{0}}} $与$ {\mathit{\boldsymbol{v}}_{\rm{0}}} $分别是基因-基因子网络和疾病-疾病子网络的初始向量,$ \eta $是赋予基因子集和表型子集的比重参数。若疾病$ {d_c} $在基因-基因子网络中有$ m $个已知疾病基因,则$ {\mathit{\boldsymbol{u}}_{\rm{0}}} $中每个疾病基因对应的分量为$ 1/m $, 其他基因对应的分量为0;$ {\mathit{\boldsymbol{v}}_{\rm{0}}} $中疾病$ {d_c} $对应的分量赋值为1,其余为0。
RWRH是典型的基于异构网络整合表型相似性信息进行疾病基因预测的方法。后续的RWRHN[64]是对RWRH的改进,其主要的创新点在于利用RWS[65]算法对背景蛋白质网络通过链路预测进行重构,从而得到一个可信度更高的PPI网络;Singh-Blom等利用Katz算法在异构网络中游走,其与RWRH的主要区别在于只考虑有限路径和异构矩阵的归一化方式不同[56];文献[66]继承和发展了Katz思想,提出HeteSim MultiPath (HSMP)方法在异构网络中衡量不同节点之间的相似性。值得注意的是基于异构网络的游走方法忽视了不同网络量级上的差别以及信息的异构性,对不同网络的转移概率矩阵采取统一处理的做法存在缺陷。漫步者能否成功地在不同网络中顺利游走且这种游走是否存在生物学意义有待进一步解释。
-
文献[61]提出的CIPHER(correlating protein interaction network and PHEnotype network to pRedict disease genes)算法定义了一个表型相似性向量$ {\mathit{\boldsymbol{S}}_{{d_c}}} $及一个基因邻近性向量$ {\mathit{\boldsymbol{ \boldsymbol{\varPhi} }}_g} $,并用这两个向量的一致性得分进行疾病基因预测。
首先构建了一个同时包含基因-基因、基因-疾病、疾病-疾病关联关系的异构网络。设此网络中共有$ n $个不同的疾病表型$ {d_1}, {d_2}, \cdots, {d_n} $以及$ m $个不同的疾病基因$ {g_1}, {g_2}, \cdots, {g_m} $,则对所研究的疾病$ {d_c} $,其表型相似性向量$ {\mathit{\boldsymbol{S}}_{{d_c}}} $定义为Minminer数据库中疾病$ {d_c} $与这$ n $个表型的表型相似分构成的向量:
$$ {{\mathit{\boldsymbol{S}}}_{{{d}_{c}}}}=({{S}_{{{d}_{c}}{{d}_{1}}}}, {{S}_{{{d}_{c}}{{d}_{2}}}}, \cdots, {{S}_{{{d}_{c}}{{d}_{n}}}}) $$ (15) 对候选基因$ g $,首先计算它与每个疾病基因在网络上的拓扑距离$ L(g, {g_j}) $$ (j = 1, 2, \cdots, m) $。CIPHER分别用两种方法计算基因间的拓扑距离,一种是直接邻居法,另一种是最短路径法。然后,对每一种疾病表型$ {d_i} $$ (i = 1, 2, \cdots, n) $,计算$ g $与种子集$ G({d_i}) $间的距离如下:
$$ {{\mathit{\Phi} }_{g{{d}_{i}}}}=\sum\limits_{{g}'\in G({{d}_{i}})}{{{\rm{e}}^{-{{L}^{2}}(g, {g}')}}} $$ (16) 从而得到基因g与所有表型的邻近性向量$ {\mathit{\boldsymbol{ \boldsymbol{\varPhi} }}_g} = ({\mathit{\Phi} _{g{d_1}}}, {\mathit{\Phi} _{g{d_2}}}, \cdots, {\mathit{\Phi} _{g{d_n}}}) $。
最后,定义一致性得分为向量$ {\mathit{\boldsymbol{S}}_{{d_c}}} $与$ {\mathit{\boldsymbol{ \boldsymbol{\varPhi} }}_g} $的皮尔逊相关系数:
$$ {\rm{C}}{{\rm{S}}_{{d_c}g}} = {\rm{corr}}({\mathit{\boldsymbol{S}}_{{d_c}}}, {\mathit{\boldsymbol{ \boldsymbol{\varPhi} }}_g}) $$ (17) 式中,$ {\rm{C}}{{\rm{S}}_{{d_c}g}} $代表候选基因$ g $与$ {d_c} $一致性得分,衡量$ g $在网络中的位置同$ {d_c} $与其他表型相似性得分的一致性,一致性得分越高越有可能是致病基因。
-
如前所述,生物学网络数据存在覆盖率不足、准确率低的缺点,以这样的数据为基础进行疾病基因预测等方面的研究,一定程度上会影响结果的准确性。目前主要采取数据融合的方法克服这一困难,这类方法可分成两种,一是对网络的融合[67],即在实施预测之前将多种组学数据利用统计推断、机器学习等方法融合为一个大网络,如FLN[26]、Hippie[30]、STRING[27]等,或者基于某个网络进行链路预测,以获得更多的潜在连接,如Biomine[68]。另一种方法则是本节介绍的融合多个结果的方法,即首先按照疾病基因预测的流程,针对不同的数据源构造不同的网络分别进行预测,最后将多种预测的结果利用统计学方法融合为最后结果。
由文献[69]开发的Endeavour是在多结果融合方面最早的研究成果。Endeavour包括两个工作阶段,第一阶段依据不同的数据源计算候选基因与致病基因之间的相似性得到候选基因的排序列表,第二阶段针对多个候选基因的排序列表,通过N维序列统计(NDOS)融合为一个最后的排序结果。尽管Endeavour相较之单个数据源的预测结果表现更好,但是仍然存在以下3个缺陷[70]:1) Endeavour对不同的数据源需要不同的衡量标准,如果想添加新的数据源,工作量将会增大且繁琐。2) 由于不同的数据源之间存在系统误差和噪音,因此在第二阶段融合多个排名时将难以衡量和消除这些误差和噪音,可能会对预测结果产生不利影响。3) Endeavour采用局部方法测量基因间的拓扑距离,预测效果不如全局方法。因此,目前有很多新方法继承和发展了Endeavour的思想,从结果融合这个方向进行疾病基因预测。
-
与Endeavour每次对单个基因分别基于单一数据源进行排名预测不同,DIR(data integration rank)同时利用多个数据源对某一基因进行排名,只采用排名最好的名次作为该基因的最终排名,即只采用对某一基因而言信息量最大的数据源作为背景网络。DIR的详细步骤如下:
1) 分别基于单个背景网络,利用扩散核计算基因对的扩散核分数K。
DIR选择扩散核算法[43]计算基因间的相似性。基因的扩散核分数越高,基因间的距离越近。
2) 基于基因对的扩散核分数计算相对重要性分值。
$$ {\rm{KP}}{{\rm{C}}^l}(i, j) = \frac{{|\{ (s, t)|{K^l}(s, t) \ge {K^l}(i, j)\} |}}{{|\{ (s, t)|{K^l}(s, t) > 0|}} $$ (18) 式中,$ l = 1, 2, \cdots, m $代表来自不同的数据源的背景网络,共有$ m $个;$ K $为扩散核距离。式中的分母表示某一背景网络中所有连接的基因对,分子表示所有连接的基因对中比基因对$ (i, j) $距离更近的基因对。直观上,$ {\rm{KP}}{{\rm{C}}^l}(i, j) $表示基因对$ (i, j) $扩散核得分的相对重要性分值,由扩散核分数大于该基因对的基因对数占总基因对数的百分比衡量。$ {\rm{KP}}{{\rm{C}}^l}(i, j) $值越小,表明在数据源$ l $中基因对$ (i, j) $之间的相似度越高。由于使用不同的网络数据源,基因在不同数据源上的拓扑距离不具有可比性,而$ {\rm{KPC}} $为在不同数据源上获得的基因$ i $与基因$ j $的扩散核分数提供了相对重要性的衡量标准。
3) 基于相对重要性分值计算最终数据融合排名$ {\rm{DIR}}(g) $,定义为:
$$ \begin{array}{c} {\rm{DIR}}(g) = \\ \frac{{\sum\limits_{a \in G({d_c})} {\max \{-\log ({\rm{KP}}{{\rm{C}}^l}(g, a), 1 \le l \le m)\} } }}{{|a \in G({d_c})|\max \{-\log ({\rm{KP}}{{\rm{C}}^l}(g, a), 1 \le l \le m)\} > 0\} |}} \end{array} $$ (19) 式中,$ g $代表候选基因;$ G({d_c}) $是疾病$ {d_c} $的已知致病基因的集合;$ {\rm{DIR}}(g) $集合了基因$ g $与所有致病基因之间的关联。每一基因对在式(18) 中基于$ m $个数据源产生了$ m $个相对重要性分值,候选基因$ g $只选取表现最好的相对重要性分值即式(19) 中分子代表$ g $与所有致病基因之间的最小的$ {\rm{KPC}} $之和。分母代表$ g $与所有致病基因之间$ {\rm{KPC}} $最小的数值,以便对数据进行归一化。由于$ {\rm{KPC}} $的分值与基因对之间的相似性成反比,因此对$ {\rm{KPC}} $取负数。$ {\rm{DIR}}(g) $得分越高,代表基因对之间相似性越大。
-
文献[70]提出了一个新的排名融合策略DRS (discounted rating system)。DRS对Endeavour的两个阶段进行改进,在第一阶段采用RWR对以单个数据源为背景网络的候选基因进行排名,在第二个阶段,基于DRS策略进行排名融合。DRS采用了4种数据源:HPRD[20]和BioGRID[72]组成的PPI网络,以及GO数据库的3个部分BP(biological process)、MF (molecular function)、CC(cellular component)分别构成的3个独立的子网络。分别基于4个网络利用RWR对候选基因打分,取前100个基因形成排名表。随后利用DRS进行排名融合,步骤如下:
1) 将排名表转化为等级表
将排名靠前的100个候选基因等分成5个等级,排名越前,等级越高。
2) 将等级表转化为打折排名表:
$$ {\rm{d}}{{\rm{r}}_i} = \frac{{{\rm{rattin}}{{\rm{g}}_i}}}{{{{\log }_2}({r_i} + 1)}} $$ (20) 式中,$ {\rm{rattin}}{{\rm{g}}_i} $是步骤一中得到的某候选基因基于数据源$ i $得到的等级;$ {r_i} $是该基因在数据源$ i $中的最初排名;$ {\rm{d}}{{\rm{r}}_i} $代表该基因在数据源$ i $中的打折排名。该方法倾向于强化排名靠前的候选基因,抑制表现较差的候选基因。
3) 基于多个背景网络融合打折排名:
$$ {S_{{\rm{dr}}}} = \frac{1}{n}\sum\limits_{i = 1}^n {{\rm{d}}{{\rm{r}}_i}} $$ (21) 对于$ n $个数据源,取打折排名的平均值作为最终的排名。实验结果表明,随着数据源数量的不断增加,DRS较之Endeavour在运行速度上有很大的优势,且获得了与其相当的AUC表现。
-
本文简单介绍疾病基因预测中常用的评价预测效果的方法,即留一交叉验证法(leave-one-out-cross-validation)、ROC曲线法、富集得分、以及模拟寻找疾病基因流程的方法。前两种方法是计算机科学中常用的检验算法优劣的方法,后两种方法是针对疾病基因预测这一特定问题的评价方法。
-
将数据源分为两类,候选基因作为测试集,已知疾病基因组成训练集。每次从训练集中选取一个疾病基因作为目标基因,将目标基因放入候选基因中组成测试集,运用算法对测试集中的每一个基因打分,验证算法是否能够成功地预测目标基因为致病基因。最后以目标基因的平均排名或者前5%或前1%作为衡量算法的预测能力的指标。候选基因一般通过一条人为的连锁区间产生,即在染色体区域选取距离目标基因最近的100个基因作为候选基因[43]。除此之外,候选基因也可以根据不同的验证目而改变。例如,文献[71]为了验证DIR算法的鲁棒性,产生了另外两组候选基因,分别是全网络的基因和随机产生的100个基因[71]。
-
在留一交互验证法中,设置$ k $为阈值,选取排名前$ k $的基因作为预测的疾病基因,称为阳性数据,在$ k $排名之后的基因认为是阴性数据。数据的属性存在以下4种情况:
1) 如果真正的阳性数据预测为阳性,则称之为真阳性数据(true positive, TP)
2) 如果真正的阴性数据被预测为阴性,则称之为真阴性数据(true negative, TN)
3) 如果真正的阳性数据被预测为阴性,则称之为假阴性数据(false negative, FN)
4) 如果真正的阴性数据被预测为阳性,则称之为假阳性数据(false positive, FP)
根据上述4种情况可以计算数据的真阳性与假阳性,ROC曲线代表着数据的假阳性与真阳性数据关系的曲线。AUC面积是ROC曲线下的面积,面积越大,算法的表现越好[74]。
-
假设有100个候选基因,如果目标基因(已知疾病基因)在打分过程中排序第一,那么该基因的富集得分为50/1;如果目标基因排名第$ n $则该基因的富集得分为$ 50/n $[26]。
-
大量研究表明,疾病基因预测倾向于那些已经得到充分研究的基因,高估了预测算法的表现。由于一旦一个致病基因被确认,会引起更多科学工作者深入研究,造成针对该基因的信息较之其他潜在基因在基因预测中更具有优势。为了避免这种知识污染,公平客观的评价算法的表现,可以模拟发现该致病基因的流程,具体如下:首先在OMIM数据库中人工地核对每个致病基因与该疾病产生关联的时间节点。假如该致病基因是在2007年以后发现的,那么将2007年以前的疾病基因作为种子集,在2007年之前的OMIM数据库中进行疾病基因预测,验证是否能成功预测出2007年之后发现的疾病基因[75]。
-
目前,基于网络的疾病基因预测方法取得了令人瞩目的发展,日益受到生物医学工作者的重视,产生了许多基于网络的疾病基因预测的在线工具[76],如Suspects[77]、ToppGene[78]、GeneDistiller[79]、GeneWander[43]、Endeavour[69]等。使用这些工具,可缩小潜在疾病基因的范围,较之传统的实验方法,极大地解放了劳动力,降低了实验耗费,减少了实验误差和系统误差。除此之外,疾病基因预测算法反过来可以作为在实验中观测到的可疑基因结果的辅助证据。越来越多的预测方法,倾向于指导非专业人员,在无需太多统计、计算方面知识的前提下,运用预测工具进行预测活动[80-81]。
尽管疾病基因预测方法在过去几年取得了巨大成就,该领域仍然存在以下挑战:1) 异构数据源的融合方法过于简单。融合后的数据缺乏足够的生物学依据,并且无法及时更新[82];2) 预测方法存在主观性。例如基于表型语义相似性的文本挖掘算法,对于表型的医学词汇描述依赖于专业人员的知识储备;3) 预测的疾病基因选取数量存在不合理性。利用算法对候选基因进行打分所得到的排名表是预测潜在致病基因的依据。常规的做法是选取前$ k $作为预测的致病基因,但是这种做法忽视了候选基因的得分显著性,可能产生较高的假阴性与假阳性。因此选取预测的致病基因应该采用更为灵活的策略,例如按照候选基因的得分趋势选取最为显著的一部分作为预测致病基因,以避免遗漏真正的候选基因;4) 预测方法和预测数据存在偏向性。基于“连接有罪”的生物学假设,一些预测方法偏向位于网络拓扑结构中心的hub节点,而忽视了那些位于网络边缘、连接度较小的节点。除此之外,不同数据源之间往往并不是相互独立的,存在交互影响,这使得数据源偏向于那些已经得到充分研究的基因;5) 预测工具的评价标准缺乏客观性。由于数据存在交互影响,对于已经发现的疾病基因存在偏向性,导致高估预测算法的表现。除此之外,目前缺少标准数据集对不同的预测工具进行无偏向的横向比较。
在基于网络的疾病基因预测领域,目前努力的方向,一是对算法进行改进以提高其预测准确性,二是用数据融合方法融合多源数据,以提高背景网络的覆盖率和可信度。除此之外,未来的疾病基因预测应当不仅仅关注候选基因在预测算法中的表现,还应当考虑候选基因的排名的$ P $值表现以及拓扑结构特征等作为辅助证据。其次,预测应当从单个基因的预测转向对蛋白质复合物和基因变异的预测,以解释更深层的疾病发病机理。最后,疾病基因预测方法也可以运用到其他生物学研究方面,例如非编码RNA、代谢物等的预测以及药物设计。尤其在药物设计方面,预测方法的创新有助于寻找药物靶标,针对不同个体制定个性化医疗。
A Survey of Disease Gene Prediction Methods Based on Molecular Networks
-
摘要: 疾病基因预测是揭示疾病作用机理、系统研究复杂疾病的关键环节。高通量生物实验技术的成熟,促进了基于分子网络的疾病基因预测方法的发展。基于“连接有罪”的生物学假设,疾病基因预测算法在生物网络中衡量候选基因与已知疾病基因的邻近性或相似性,以预测潜在的致病基因。该文将疾病基因预测方法归纳为3种:基于已知疾病基因信息的预测方法、融合表型相似性信息的预测方法以及融合多结果的预测方法,并对这3种方法的研究现状进行了综述,指出了现有研究成果的不足以及未来的研究方向。Abstract: The identification of disease genes is the crucial step in uncovering disease pathology and systematically analyzing polygenetic disease. The high-throughput technology has advanced the development of network-based approaches for disease gene prediction. Based on the "guilt-by-association" principle, now disease gene prioritization methods can measure the proximity between candidate genes and causal genes so as to pinpoint the potential disease genes. In this review, we first classify the network-based approaches for disease gene prediction into three categories:the approach based on disease genes information, the approach integrated with phenotype similarity and the approach that integrates several results from multiple data resources into one final result. Then we bring out the current situation of these approaches and summarize the current achievements and existing problems. Finally we put forward some suggestions for future research.
-
[1] LAN W, WANG J, LI M, et al. Computational approaches for prioritizing candidate disease genes based on PPI networks[J]. Tsinghua Science and Technology, 2015, 20(5):500-512. doi: 10.1109/TST.2015.7297749 [2] EASTON D, BISHOP D, FORD D, et al. Genetic linkage analysis in familial breast and ovarian cancer:Results from 214 families the breast cancer linkage consortium[J]. American Journal of Human Genetics, 1993, 52(4):678. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1682082/ [3] OTT J, WANG J, LEAL S M. Genetic linkage analysis in the age of whole-genome sequencing[J]. Nature Reviews Genetics, 2015, 16(5):275-284. doi: 10.1038/nrg3908 [4] GOH K-I, CUSICK M E, VALLE D, et al. The human disease network[J]. Proceedings of the National Academy of Sciences, 2007, 104(21):8685-8690. doi: 10.1073/pnas.0701361104 [5] BRUNNER H G, VAN DRIEL M A. From syndrome families to functional genomics[J]. Nature Reviews Genetics, 2004, 5(7):545-551. doi: 10.1038/nrg1383 [6] LAGE K, KARLBERG E O, STØRLING Z M, et al. A human phenome-interactome network of protein complexes implicated in genetic disorders[J]. Nature Biotechnology, 2007, 25(3):309-316. doi: 10.1038/nbt1295 [7] BARABÁSI A-L, GULBAHCE N, LOSCALZO J. Network medicine:a network-based approach to human disease[J]. Nature Reviews Genetics, 2011, 12(1):56-68. doi: 10.1038/nrg2918 [8] TIFFIN N, ANDRADE-NAVARRO M A, PEREZIRATXETA C. Linking genes to diseases:it's all in the data[J]. Genome Medicine, 2009, 1(8):77. doi: 10.1186/gm77 [9] ANTANAVICIUTE A, DALY C, CRINNION L A, et al. GeneTIER:Prioritization of candidate disease genes using tissue-specific gene expression profiles[J]. Bioinformatics, 2015, 31(16):2728-2735. doi: 10.1093/bioinformatics/btv196 [10] CRUZ-MONTEAGUDO M, BORGES F, PAZ-Y-MIÑO C, et al. Efficient and biologically relevant consensus strategy for Parkinson's disease gene prioritization[J]. BMC Medical Genomics, 2016, 9(1):12. doi: 10.1186/s12920-016-0173-x [11] RUAL J-F, VENKATESAN K, HAO T, et al. Towards a proteome-scale map of the human protein-protein interaction network[J]. Nature, 2005, 437(7062):1173-1178. doi: 10.1038/nature04209 [12] TEJERA E, BERNARDES J, REBELO I. Co-expression network analysis and genetic algorithms for gene prioritization in preeclampsia[J]. BMC Medical Genomics, 2013, 6(1):51. doi: 10.1186/1755-8794-6-51 [13] CARTER S L, BRECHBÜHLER C M, GRIFFIN M, et al. Gene co-expression network topology provides a framework for molecular characterization of cellular state[J]. Bioinformatics, 2004, 20(14):2242-2250. doi: 10.1093/bioinformatics/bth234 [14] NITSCH D, GONÇALVES J P, OJEDA F, et al. Candidate gene prioritization by network analysis of differential expression using machine learning approaches[J]. BMC Bioinformatics, 2010, 11(1):460. doi: 10.1186/1471-2105-11-460 [15] LI M, LI Q, GANEGODA G U, et al. Prioritization of orphan disease-causing genes using topological feature and GO similarity between proteins in interaction networks[J]. Science China Life Sciences, 2014, 57(11):1064-1071. doi: 10.1007/s11427-014-4747-6 [16] SCHLICKER A, LENGAUER T, ALBRECHT M. Improving disease gene prioritization using the semantic similarity of Gene Ontology terms[J]. Bioinformatics, 2010, 26(18):i561-i567. doi: 10.1093/bioinformatics/btq384 [17] OLIVER S. Proteomics:Guilt-by-association goes global[J]. Nature, 2000, 403(6770):601-603. doi: 10.1038/35001165 [18] OTIM, BRUNNER H G. The modular nature of genetic diseases[J]. Clinical Genetics, 2007, 71(1):1-11. doi: 10.1111/j.1399-0004.2006.00708.x/abstract [19] CAGNEY G, UETZ P, FIELDS S. High-throughput screening for protein-protein interactions using two-hybrid assay[J]. Methods in Enzymology, 2000, 328:3-14. doi: 10.1016/S0076-6879(00)28386-9 [20] PRASAD T S K, GOEL R, KANDASAMY K, et al. Human protein reference database-2009 update[J]. Nucleic Acids Research, 2009, 37(suppl 1):D767-D772. doi: 10.1093/nar/gkn892 [21] CHATR-ARYAMONTRI A, BREITKREUTZ B-J, OUGHTRED R, et al. The BioGRID interaction database:2015 update[J]. Nucleic Acids Research, 2015, 43(D1):D470-D478. doi: 10.1093/nar/gku1204 [22] BADER G D, BETEL D, HOGUE C W. BIND:the biomolecular interaction network database[J]. Nucleic Acids Research, 2003, 31(1):248-250. doi: 10.1093/nar/gkg056 [23] LICATA L, BRIGANTI L, PELUSO D, et al. MINT, the molecular interaction database:2012 update[J]. Nucleic Acids Research, 2012, 40(D1):D857-D861. doi: 10.1093/nar/gkr930 [24] KERRIEN S, ARANDA B, BREUZA L, et al. The IntAct molecular interaction database in 2012[J]. Nucleic Acids Research, 2011, 40(D1):D841-D846. http://europepmc.org/articles/PMC3245075 [25] AMARAL L A N. A truer measure of our ignorance[J]. Proceedings of the National Academy of Sciences, 2008, 105(19):6795-6796. doi: 10.1073/pnas.0802459105 [26] LINGHU B, SNITKIN E S, HU Z, et al. Genome-wide prioritization of disease genes and identification of disease-disease associations from an integrated human functional linkage network[J]. Genome Biology, 2009, 10(9):91. doi: 10.1186/gb-2009-10-9-r91 [27] SZKLARCZYK D, FRANCESCHINI A, WYDER S, et al. STRING v10:Protein-protein interaction networks, integrated over the tree of life[J]. Nucleic Acids Research, 2014, 43(D1):D447-D452. https://academic.oup.com/nar/article/43/D1/D447/2435295/STRING-v10-protein-protein-interaction-networks [28] LEE I, BLOM U M, WANG P I, et al. Prioritizing candidate disease genes by network-based boosting of genome-wide association data[J]. Genome Research, 2011, 21(7):1109-1121. doi: 10.1101/gr.118992.110 [29] SCHMITT T, OGRIS C, SONNHAMMER E L. FunCoup 30:Database of genome-wide functional coupling networks[J]. Nucleic Acids Research, 2014, 42(D1):D380-D388. doi: 10.1093/nar/gkt984 [30] SCHAEFER M H, FONTAINE J-F, VINAYAGAM A, et al. HIPPIE:Integrating protein interaction networks with experiment based quality scores[J]. PloS One, 2012, 7(2):e31826. doi: 10.1371/journal.pone.0031826 [31] MOREAU Y, TRANCHEVENT L-C. Computational tools for prioritizing candidate genes:Boosting disease gene discovery[J]. Nature Reviews Genetics, 2012, 13(8):523-536. doi: 10.1038/nrg3253 [32] HAMOSH A, SCOTT A F, AMBERGER J S, et al. Online mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders[J]. Nucleic Acids Research, 2005, 33(suppl 1):D514-D517. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC99152/ [33] BECKER K G, BARNES K C, BRIGHT T J, et al. The genetic association database[J]. Nature Genetics, 2004, 36(5):431-432. doi: 10.1038/ng0504-431 [34] PENNISI E. Europe's cancer genome anatomy project[J]. Science, 1997, 276(5315):1024. doi: 10.1126/science.276.5315.1024 [35] FUTREAL P A, COIN L, MARSHALL M, et al. A census of human cancer genes[J]. Nature Reviews Cancer, 2004, 4(3):177-183. doi: 10.1038/nrc1299 [36] BAUER-MEHREN A, RAUTSCHKA M, SANZ F, et al. DisGeNET:a cytoscape plugin to visualize, integrate, search and analyze gene-disease networks[J]. Bioinformatics, 2010, 26(22):2924-2926. doi: 10.1093/bioinformatics/btq538 [37] FREUDENBERG J, PROPPING P. A similarity-based method for genome-wide prediction of disease-relevant human genes[J]. Bioinformatics, 2002, 18(suppl 2):S110-S115. doi: 10.1093/bioinformatics/18.suppl_2.S110 [38] VAN DRIEL M A, BRUGGEMAN J, VRIEND G, et al. A text-mining analysis of the human phenome[J]. European Journal of Human Genetics, 2006, 14(5):535-542. doi: 10.1038/sj.ejhg.5201585 [39] VAN DRIEL M A, BRUGGEMAN J, VRIEND G, et al. MimMiner:a online mendelian inheritance in man mining tool[DB/OL].[2006-05-08]. http://wwwcmbirunl/MimMiner/supplhtml. [40] OTIV M, SNEL B, HUYNEN M A, et al. Predicting disease genes using protein-protein interactions[J]. Journal of Medical Genetics, 2006, 43(8):691-698. doi: 10.1136/jmg.2006.041376 [41] KRAUTHAMMER M, KAUFMANN C A, GILLIAM T C, et al. Molecular triangulation:Bridging linkage and molecular-network information for identifying candidate genes in Alzheimer's disease[J]. Proceedings of the National Academy of Sciences of the United States of America, 2004, 101(42):15148-15153. doi: 10.1073/pnas.0404315101 [42] NAVLAKHA S, KINGSFORD C. The power of protein interaction networks for associating genes with diseases[J]. Bioinformatics, 2010, 26(8):1057-1063. doi: 10.1093/bioinformatics/btq076 [43] KÖHLER S, BAUER S, HORN D, et al. Walking the interactome for prioritization of candidate disease genes[J]. The American Journal of Human Genetics, 2008, 82(4):949-958. doi: 10.1016/j.ajhg.2008.02.013 [44] VANUNU O, MAGGER O, RUPPIN E, et al. Associating genes and protein complexes with disease via network propagation[J]. PLoS Comput Biol, 2010, 6(1):e1000641. doi: 10.1371/journal.pcbi.1000641 [45] KATZ L. A new status index derived from sociometric analysis[J]. Psychometrika, 1953, 18(1):39-43. doi: 10.1007/BF02289026 [46] ZHANG S, NING X M, ZHANG X S. Graph kernels, hierarchical clustering, and network community structure:experiments and comparative analysis[J]. The European Physical Journal B-Condensed Matter and Complex Systems, 2007, 57(1):67-74. doi: 10.1140/epjb/e2007-00146-y [47] FOUSS F, PIROTTE A, RENDERS J-M, et al. Random-walk computation of similarities between nodes of a graph with application to collaborative recommendation[J]. IEEE Transactions on Knowledge and Data Engineering, 2007, 19(3):355-369. doi: 10.1109/TKDE.2007.46 [48] TAUCHEN G. Finite state markov-chain approximations to univariate and vector autoregressions[J]. Economics Letters, 1986, 20(2):177-181. doi: 10.1016/0165-1765(86)90168-0 [49] BRIN S, PAGE L. Reprint of:the anatomy of a large-scale hypertextual web search engine[J]. Computer Networks, 2012, 56(18):3825-3833. doi: 10.1016/j.comnet.2012.10.007 [50] 汪小帆, 李翔, 陈关荣.网络科学导论[M].北京:高等教育出版社, 2012. WANG Xiao-fan, LI Xiang, CHEN Guan-rong. Network science:an introduction[M]. Beijing:Higher Education Press, 2012. [51] LIU W, LÜ L. Link prediction based on local random walk[J]. EPL (Europhysics Letters), 2010, 89(5):58007. doi: 10.1209/0295-5075/89/58007 [52] 吕琳媛, 周涛.链路预测[M].北京:高等教育出版社, 2013:69-70. LÜ Lin-yuan, ZHOU Tao. Link Prediction[M]. Beijing:Higher Education Press, 2013:69-70. [53] LÜ L, ZHOU T. Link prediction in complex networks:a survey[J]. Physica A:Statistical Mechanics and Its Applications, 2011, 390(6):1150-1170. doi: 10.1016/j.physa.2010.11.027 [54] 吕琳媛.复杂网络链路预测[J].电子科技大学学报, 2010, 39(5):651-661. http://youxian.cnki.com.cn/yxdetail.aspx?filename=WJFZ20170801015&dbname=CAPJ2015 LÜ Lin-yuan. Link prediction on complex networks[J]. Journal of University of Electronic Science and Technology of China, 2010, 39(5):651-661. http://youxian.cnki.com.cn/yxdetail.aspx?filename=WJFZ20170801015&dbname=CAPJ2015 [55] ZHAO J, YANG T H, HUANG Y, et al. Ranking candidate disease genes from gene expression and protein interaction:a Katz-centrality based approach[J]. PloS One, 2011, 6(9):e24306. doi: 10.1371/journal.pone.0024306 [56] SINGH-BLOM U M, NATARAJAN N, TEWARI A, et al. Prediction and validation of gene-disease associations using methods inspired by social network analyses[J]. PloS One, 2013, 8(5):e58977. doi: 10.1371/journal.pone.0058977 [57] ERTEN S, BEBEK G, EWING R M, et al. DADA:Degree-aware algorithms for network-based disease gene prioritization[J]. BioData Mining, 2011, 4(1):19. doi: 10.1186/1756-0381-4-19 [58] WAGNER G P, PAVLICEV M, CHEVERUD J M. The road to modularity[J]. Nature Reviews Genetics, 2007, 8(12):921-931. doi: 10.1038/nrg2267 [59] OTI M, HUYNEN M A, BRUNNER H G. Phenome connections[J]. Trends in Genetics, 2008, 24(3):103-106. doi: 10.1016/j.tig.2007.12.005 [60] ERTEN S, BEBEK G, KOYUTÜRK M. Vavien:an algorithm for prioritizing candidate disease genes based on topological similarity of proteins in interaction networks[J]. Journal of Computational Biology, 2011, 18(11):1561-1574. doi: 10.1089/cmb.2011.0154 [61] WU X, JIANG R, ZHANG M Q, et al. Network-based global inference of human disease genes[J]. Molecular Systems Biology, 2008, 4(1):189. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2424293/ [62] GANEGODA G U, SHENG Y, WANG J. ProSim:a method for prioritizing disease genes based on protein proximity and disease similarity[J]. BioMed Research International, 2015(5):213750. http://europepmc.org/articles/PMC4538409 [63] LI Y, PATRA J C. Genome-wide inferring genephenotype relationship by walking on the heterogeneous network[J]. Bioinformatics, 2010, 26(9):1219-1224. doi: 10.1093/bioinformatics/btq108 [64] LUO J, LIANG S. Prioritization of potential candidate disease genes by topological similarity of protein-protein interaction network and phenotype data[J]. Journal of Biomedical Informatics, 2015, 53:229-236. doi: 10.1016/j.jbi.2014.11.004 [65] LEI C, RUAN J. A novel link prediction algorithm for reconstructing protein-protein interaction networks by topological similarity[J]. Bioinformatics, 2013, 29(3):355-364. doi: 10.1093/bioinformatics/bts688 [66] ZENG X, LIAO Y, ZOU Q. Prediction and validation of disease genes using HeteSim scores[J]. IEEE/ACM Transactions on Computational Bilolgy and Bioinformatics, 2017, 14(3):687-695. doi: 10.1109/TCBB.2016.2520947 [67] BERSANELLI M, MOSCA E, REMONDINI D, et al. Methods for the integration of multi-omics data:Mathematical aspects[J]. BMC Bioinformatics, 2016, 17(2):167. doi: 10.1186/s12859-015-0857-9 [68] ERONEN L, TOIVONEN H. Biomine:Predicting links between biological entities using network models of heterogeneous databases[J]. BMC Bioinformatics, 2012, 13(1):119. doi: 10.1186/1471-2105-13-119 [69] AERTS S, LAMBRECHTS D, MAITY S, et al. Gene prioritization through genomic data fusion[J]. Nature Biotechnology, 2006, 24(5):537-544. doi: 10.1038/nbt1203 [70] LI Y, PATRA J C. Integration of multiple data sources to prioritize candidate genes using discounted rating system[J]. BMC Bioinformatics, 2010, 11(1):S20. doi: 10.1186/1471-2105-11-20 [71] CHEN Y, WANG W, ZHOU Y, et al. In silico gene prioritization by integrating multiple data sources[J]. PloS One, 2011, 6(6):e21137. doi: 10.1371/journal.pone.0021137 [72] STARK C, BREITKREUTZ B-J, CHATRARYAMONTRI A, et al. The BioGRID interaction database:2011 update[J]. Nucleic Acids Research, 2011, 39(suppl 1):D698-D704. https://academic.oup.com/nar/article/39/suppl_1/D698/2507010/The-BioGRID-Interaction-Database-2011-update [73] ZOU Q, LI J, WANG C, et al. Approaches for recognizing disease genes based on network[J]. BioMed Research International, 2014(5013):416323. https://www.hindawi.com/journals/bmri/2014/416323/ [74] FAWCETT T. An introduction to ROC analysis[J]. Pattern Recognition Letters, 2006, 27(8):861-874. doi: 10.1016/j.patrec.2005.10.010 [75] BÖRNIGEN D, TRANCHEVENT L-C, BONACHELACAPDEVILA F, et al. An unbiased evaluation of gene prioritization tools[J]. Bioinformatics, 2012, 28(23):3081-3088. doi: 10.1093/bioinformatics/bts581 [76] TRANCHEVENT L-C, CAPDEVILA F B, NITSCH D, et al. A guide to web tools to prioritize candidate genes[J]. Briefings in Bioinformatics, 2011, 12(1):22-32. doi: 10.1093/bib/bbq007 [77] ADIE E A, ADAMS R R, EVANS K L, et al. SUSPECTS:Enabling fast and effective prioritization of positional candidates[J]. Bioinformatics, 2006, 22(6):773-774. doi: 10.1093/bioinformatics/btk031 [78] CHEN J, XU H, ARONOW B J, et al. Improved human disease candidate gene prioritization using mouse phenotype[J]. BMC Bioinformatics, 2007, 8(1):392. doi: 10.1186/1471-2105-8-392 [79] SEELOW D, SCHWARZ J M, SCHUELKE M. GeneDistiller-distilling candidate genes from linkage intervals[J]. PLoS One, 2008, 3(12):e3874. doi: 10.1371/journal.pone.0003874 [80] CASCI T. Human disease:Something old, something new[J]. Nature Reviews Genetics, 2011, 12(6):382-383. http://nas-sites.org/ilarjournal/previous-issues/volume-52-number-1/introduction-animal-models-of-aging-something-old-something-new/ [81] HUANG D W, SHERMAN B T, LEMPICKI R. A systematic and integrative analysis of large gene lists using DAVID bioinformatics resources[J]. Nature Protocols, 2009, 4(1):44-57. https://www.ncbi.nlm.nih.gov/pubmed/19131956 [82] WANG X, GULBAHCE N, YU H. Network-based methods for human disease gene prediction[J]. Briefings in Functional Genomics, 2011, 10(5):280-293. doi: 10.1093/bfgp/elr024 -
 点击查看大图
点击查看大图
图(1)
计量
- 文章访问数: 4661
- HTML全文浏览量: 1336
- PDF下载量: 171
- 被引次数: 0
