 ISSN
ISSN 




-
材料辐照损伤是当前材料领域和计算机领域研究的热点之一。在辐照条件下的材料模拟中,入射粒子与材料中的晶格原子相互碰撞并传递能量,晶格原子获得能量后将离开晶格点阵并引发级联碰撞,形成点缺陷及团簇。在辐射条件下,点缺陷将复合、迁移、聚集成团,最终导致材料微观结构演化以及宏观力学性能退化。多尺度模拟是研究材料辐照效应的有效手段,可从不同的时间、空间尺度研究材料的损伤机理,预测材料的损伤程度。对材料辐照损伤的研究是一个跨越飞秒和年的大时间尺度以及跨越纳米和米的大空间尺度问题。实现从原子尺度到宏观尺度的耦合模拟,是一个庞大而复杂的工程,而每一个尺度又可划分为多个更细的尺度,分别对应不同的模拟方法。本文只研究微观多尺度上的耦合模拟。在微观尺度上,分子动力学(MD)和动力学蒙特卡洛(KMC)为常用的两种经典的材料模拟方法。MD方法[1]通过求解动量守恒、质量守恒和能量守恒这三大守恒定律中的运动方程,来描述系统某一时刻的状态,再通过积分运算获得系统的性质。KMC方法[2]是一种基于概率统计理论的方法,通过计算事件的发生概率和使用随机数获得系统组态的跃迁。由于KMC方法的计算对象是组态而非原子,对原子的特征描述维度(如:速度、力、质量等)比MD少,故其模拟的时间尺度和空间尺度都比MD大。虽然,MD方法对系统的描述比KMC方法更加准确,但由于模拟的时间步长太短(约为10-15 s),使得计算量和计算开销巨大,对计算机的运行性能要求也较高[3]。与之相反,KMC方法则可突破MD方法在时间尺度上的限制,可达到秒或者更长的模拟时间,此方法可以对系统的动力学特征进行描述,但由于它使用随机数而降低了对系统描述的准确性。因此,目前常用的做法是将MD方法和KMC方法耦合起来,在微观尺度模拟材料辐照损伤中缺陷的形成及演化过程。
多尺度模拟的关键之一在于各层次之间信息传递的准确性,同一系综下多尺度模拟方法可以实现信息的传递,提高模拟精度。多尺度模拟方法在材料辐照损伤研究方面已开展大量工作,并取得了很好的结果,如文献[4]针对材料辐照损伤问题,开发了预测性计算工具来模拟辐照材料的微观结构变化。描述了如何使用来自数千个MD模拟的碰撞级联的统计平均值来为KMC模拟提供输入,该模拟可以处理更大的尺寸,更多的缺陷和更长的持续时间。文献[5]结合分子动力学方法和动力学蒙特卡罗方法,研究了单个粒子入射硅引起的位移损伤缺陷的产生和演化过程。文献[6]提出了自适应动力学蒙特卡洛方法,该方法将KMC方法的简单性与基于MD的鞍点搜索算法相结合来模拟亚稳系统。文献[7]拓展了多尺度耦合模拟工具MaMiCo,该工具将连续流体动力学求解器与离散粒子动力学耦合,引入了对分子动力学模拟集合的采样,最后通过使用超级计算机Shaheen Ⅱ的多达65 536个计算核心来验证了分子连续模拟的并行可扩展性。文献[8]采用KMC获得富铜析出物,然后将KMC的模拟结果传给相场,用相场方法模拟富铜析出物的长大过程,同时采用MD方法对材料的结构进行检查,确保模拟过程中材料结构的一致性,有效地模拟了铁中富铜析出物的粗化过程。文献[9]通过MD的模拟和KMC顺序耦合的方法(MD的模拟结果作为KMC模拟的输入)分别模拟了铜和铁的辐照损伤过程。文献[10]利用MD方法计算系统的能量,将计算获得的能量用于KMC计算来研究铁中氦-空位团簇的增长和收缩机制。
本文通过对动力学方法和动力学蒙特卡洛方法的特点分析,将分子动力学方法和动力学蒙特卡洛方法进行耦合,可进一步突破现有时空尺度的限制,从而实现长时间、多空间尺度的原子层次直接模拟。
-
本文中,MD和KMC分别模拟材料辐照损伤的两个阶段,MD模拟缺陷的产生及团簇的形成过程为KMC模拟提供初始的原子排布和缺陷信息,KMC模拟完成缺陷从小到大演化的模拟过程。
-
MD方法[1]的基本思想是把所有物质都看成是由电子、原子或分子组成的粒子系统,每个粒子在其他粒子所提供的经验势场的作用下运动,这种运动被认为遵循牛顿运动定律。若已知体系中各粒子的初始位置,通过势能函数计算出粒子所对应的势能,利用势能求得粒子在体系中所受的力,再通过求解牛顿运动方程,计算出粒子的加速度,从而可以通过对时间的积分计算得到粒子在任意时刻的速度以及位置。如果时间足够长,即经过的循环迭代次数足够多,最终获得材料的宏观性能就越明显。
KMC模拟方法[2]是一种通过构造随机过程来模拟体系长时间演化的方法。KMC一般用来模拟由组态跃迁主导的体系的长时间(通常是秒或者更长)的演化。
本文中的KMC模拟程序采用最常用的一种KMC算法直接法(direct method),效率非常高,属于无拒绝KMC。每一步只需要产生两个平均分布在(0, 1]之间的随机数r1和r2。其中,r1被用来选定跃迁途径,r2确定模拟的前进时间。设体系处于态i,将每条跃迁途径j想象成长度与跃迁速率kij成正比的线段。将这些线段首尾相连,如果r1×ktotal落在线段jk中,则这个线段所代表的跃迁途径jk就被选中,体系移动到态jk,同时由随机数r2计算体系前进时间。
-
由于MD和KMC这两种方法的计算原理和过程不同,所关注的原子属性也不一样。其中,在MD方法中原子的位置(三维坐标)可以为模拟空间中的任意位置。而在KMC模拟中,由于KMC对间隙原子的跃迁情况计算复杂、耗时较多,在目前的系统中暂时不对间隙原子做KMC模拟。且由于KMC关注的是体系从一个状态到另一个状态的“跃迁”,故在KMC的模拟体系中,原子位置为其晶格的格点位置,原子不会出现在晶格中除格点外的其他位置上。因为存在上述这些不同,所以,在MD模拟结束后,MD模拟为KMC模拟提供的原子排布和缺陷信息无法在KMC程序中直接使用。为了解决这一问题,且遵循软件开发高内聚低耦合准则,本文开发了一个基于MD和KMC耦合模拟的耦合程序。它的主要功能是过滤间隙原子,实现原子位置重置(置于对应格点),以及完成数据的格式转换工作。MD和KMC耦合模拟的示意图如图 1所示。其中,t1为MD模拟的当前累计模拟时间,tMD为MD模拟的总时间,t2为KMC模拟的当前累计模拟时间,tKMC为KMC模拟的总时间。

图 1 MD和KMC耦合模拟示意图
-
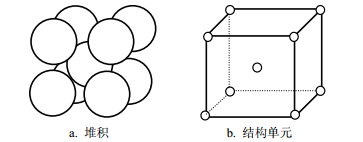
要想实现该耦合程序,必须要知道体系中数据所表示的含义,了解模拟对象的晶格结构和空间排布。本文研究是以铁为基质,混有少量铜杂质的金属合金RPV钢(RPV钢中的杂质除了铜,还有镍、锰等,由于其他杂质的含量少于铜,因此本文只研究铜杂质在辐照损伤中对RPV钢的影响)。虽然,纯铜固体的晶格结构为面心立方晶格(face-centered cubic, FCC)结构,但由于合金中铜含量极少,且在研究范围内的铜团簇半径很小,因此整个体系仍是体心立方晶格(body-centered cubic, BCC)结构。图 2就是BCC的结构示意图,圆球表示格点。

图 2 体心立方晶格的堆积与结构单元
图 2a展示的就是体心立方晶格的一个堆积,图 2b为对应的结构单元,圆球所在的位置是晶格点位置。当出现多个原子对应一个格点时,只有一个原子是正常原子,其他原子称为间隙原子。如果格点上没有原子,则该格点为空位。如有间隙原子出现,则在体系内必然存在同样数目的空位,空位和间隙原子都属于缺陷。在MD模拟中原子可以出现在晶格的任意位置,而在KMC模拟中原子都在圆点位置,不会发生偏移。因此,要想实现MD和KMC的顺序耦合,首先必须确定MD中每个原子属于哪个格点,并进行原子位置重置,将原子置于对应的格点位置。
目前,在MD的模拟中可以处理间隙原子,但在KMC模拟中不能处理间隙原子(计算太过复杂),所以MD和KMC耦合模拟的耦合程序要对所有原子进行过滤。需要注意的是,由于杂质原子在模拟过程中对结果的影响较大,所以在处理除去间隙原子的过程中本文采取杂质原子优先保留的策略,即如果一个格点同时对应一个铜原子和一个铁原子,将会选择铁原子为间隙原子,并将其从体系中除去。
由上可知,原子的位置重置和间隙原子过滤都需要先判定原子属于哪个格点。如何确定原子对应的格点,以及判定原子的类型(间隙原子、空位或正常原子)是耦合程序实现中的难点。在第2章中将详细介绍这些问题的解决方法。
-
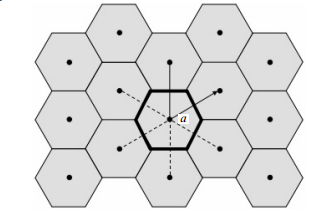
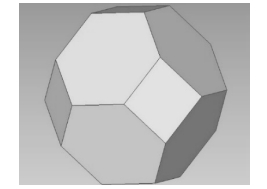
判断偏离格点位置的原子属于哪个格点是实现原子位置重置和过滤间隙原子的关键。在可视化软件OVITO[11]中就采用了Wigner-Seitz原胞缺陷分析法对可视化的原子进行分析。Wigner-Seitz原胞是晶格中比较对称的一种原胞,以一个格点为原点,作原点与其他格点连接的中垂面(二维空间中为中垂线),由这些中垂面(二维空间中为中垂线)所围成的最小体积(二维空间中为面积),图 3和图 4分别为在二维空间和三维空间中BCC结构材料的Wigner-Seitz原胞。其中,图 3中的加粗区域为格点的Wigner-Seitz原胞区域,该区域为等边六边形;图 4中每个格点的Wigner-Seitz原胞区域为一个截角八面体。

图 3 二维空间中BCC结构材料的Wigner-Seitz原胞

图 4 三维空间中BCC结构材料的Wigner-Seitz原胞
Wigner-Seitz原胞缺陷分析法的思想是:由于每个Wigner-Seitz原胞中只包含一个原子,即它的原点处的格点对应的原子如果出现多个,则其他原子就是间隙原子,就是MD和KMC中间处理过程需过滤掉的原子。如果某个Wigner-Seitz原胞中不含任何原子,则对应格点即为空位。在材料模拟和分析中通常也将空位看成是一种特殊的原子。
-
可视化软件OVITO[11]的处理过程需要初始原子位置文件和分析时刻的原子位置文件这两个文件,通过位置对照,利用Wigner-Seitz原胞缺陷分析法对要分析的原子中的缺陷进行识别。考虑到在MD和KMC耦合过程中若用OVITO中的方法实现Wigner-Seitz原胞缺陷分析法,需要存储体系的初始原子分布,且本文的研究对象为BCC结构的固体材料,原子的原始排布(完美晶格)已确定,故本文依据Wigner-Seitz原胞法[11]的原理,提出了通过距离判断寻找原子对应晶格点的算法,以此来实现间隙原子、空位和正常原子的判定,该算法被称为最短距离(SD)算法。在SD算法中不需要存储系统的初始原子分布,从而大大节约了内存开销。
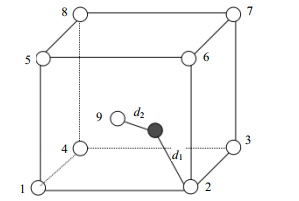
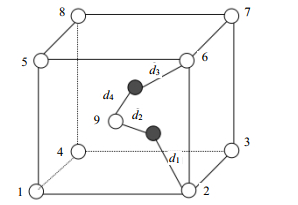
在整个物理空间中,距离任意一点最近的格点为其所在的Wigner-Seitz原胞对应的原点。因此,SD算法的核心思想就是利用距离找出距离该原子最近的格点即可。图 5为MD模拟中的原子处于非标准格点处的情况,本文给每个格点位置(空心圆圈)编号为1、2、3、4、5、6、7、8、9,其中实心圆圈表示原子。

图 5 MD模拟中原子处于非标准格点处的情况
采用SD算法计算原子与格点的映射关系的过程如下:体系中的每个原子都一定处于某个确定的晶格中,则距离原子最近的格点必定是它所在的晶格立方体的体心处格点或顶点处的格点(简称为体心格点、顶点格点)。若某个顶点格点在各个维度上的坐标与对应的原子坐标相减的绝对值都是最小,则该格点为距离原子最近的顶点格点(假设为图 5中的2号格点),计算两者间的距离设为d1;然后计算晶格中的体心格点与原子的距离设为d2;如果d1 > d2,则原子对应的格点为9号格点(体心格点),否则对应的格点为2号格点(顶点格点)。如果有多个原子同时对应9号格点,则分为两种情况处理:一种情况是多个原子中有铁原子也有两个以上杂质原子,此时将铁原子全部视为间隙原子。按照遍历顺序,将第一个杂质原子保留,其他视为间隙原子。另一种情况,只有铁原子或只有杂质原子,则按照遍历顺序,保留第一个处理的原子,其他都视为间隙原子。图 6所示为两个原子同时对应9号格点的情况,即d3 > d4且d1 > d2。

图 6 一个格点(9号)同时对应两个原子的情况
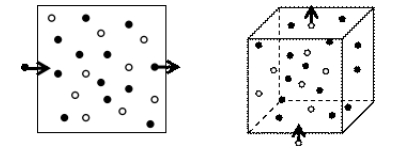
在正则系综下为了保证整个体系中模拟的原子数目不变,在MD模拟和KMC模拟中都采用了周期性边界条件(periodic boundary conditions, PBC)[12]来处理模拟盒边界上原子在空间里的运动问题。在处理原子与格点映射以及除去原子的过程中,周期性边界条件也是必须要考虑的一个问题。在整个耦合模拟的过程中,模拟盒的大小是固定不变的,采用周期性边界条件使得在原子运动过程中当有一个或几个原子从模拟盒的边界跑出时,就从相反的界面进入一个或几个原子,从而保证在体系中原子的数目是恒定的。图 7分别为二维空间和三维空间中周期性边界条件的处理情况。其中,黑色点和白色点分别表示两种不同的原子,正方形和正方体用来表示模拟盒。算法1为SD算法的伪代码实现。

图 7 二维空间和三维空间中的周期性边界条件示意图
算法1 最短距离算法
Input:模拟盒大小、晶格常数、原子坐标
Output: input_KMC.xyz,interstitialatom.xyz
Dim vertex[n1][n2][n3] center[n1][n2][n3] d1 d2 As INTEGER
//定义顶点格点计数变量与体心格点计数变量
//定义原子分别到顶点格点的距离和体心格点的距离
//n1、n2、n3分别为体系x、y、z方向上的晶胞数(BCC材料每个晶胞包含一个顶点格点和一个体心格点)。
// input_KMC.xyz为KMC的输入,内容为体系的格点坐标和对应的原子类型;
// interstitialatom.xyz为过滤出的间隙原子的坐标;
Begin:
Read 模拟盒大小、晶格常数、原子坐标
For all 铜原子(x, y, z)
计算与原子最近的顶点格点(xv, yv, zv)之间的距离d1;
计算与原子最近的体心格点(xc, yc, zc)之间的距离d2;
If d1 > d2 then
If center[xc][yc][zc] > 0 then
将铜原子(x, y, z)的信息输出到interstitialatom.xyz;
Else
center[xc][yc][zc]= center[xc][yc][zc]+1;
将原子类型、格点坐标信息输出到input_KMC.xyz;
End if
Else
If vertex [xv][yv][zv] > 0 then
将铜原子(x, y, z)的信息输出到interstitialatom.xyz;
Else
vertex [xv][yv][zv]=vertex [xv][yv][zv]+1;
将原子类型、格点坐标信息输出到input_KMC.xyz;
End if
End if
End for
For all铁原子(x, y, z)
计算与原子最近的顶点格点(xv, yv, zv)之间的距离d1;
计算与原子最近的体心格点(xc, yc, zc)之间的距离d2;
If d1 > d2 then
If center[xc][yc][zc] > 0 then
将铁原子(x, y, z)的信息输出到interstitialatom.xyz;
Else
center[xc][yc][zc]= center[xc][yc][zc]+1;
将原子类型、格点坐标信息输出到input_KMC.xyz;
End if
Else
If vertex [xv][yv][zv] > 0 then
将铁原子(x, y, z)的信息输出到interstitialatom.xyz;
Else
vertex [xv][yv][zv]= vertex [xv][yv][zv]+1;
将原子类型、格点坐标信息输出到input_KMC.xyz;
End if
End if
End for
Output格点的坐标及格点的原子类型
End
-
本文中的所有测试都是在新一代超级计算集群“元-Era”上进行的。该集群总共拥有270台曙光CB60-G16双路刀片,CPU整体性能达到120.96 T flops。每台刀片计算节点配置2个Intel E5-2680 V2(Ivy Bridge|10 C|2.8 GHz)处理器和64 GB DDR3 ECC 1866 MHz内存,每个处理器含10个处理核心。操作系统为RedHat Linux 4.4.7-3版本,编译器为Intel C++ 2013_sp1版,MPI环境为MVAPCH2-intel1.9版本。中间程序的编程语言为C语言,不需要其他外部库的支持,可在任何一台具有C编译器的linux服务器上运行。
-
为了验证本文提出的SD算法实现Wigner-Seitz原胞缺陷分析的正确性,本文使用最短距离法处理后获得的结果与OVITO软件的处理结果进行了对比。由于SD算法中在处理多个原子映射到同一个格点时,采取了杂质原子优先保留和同等条件下先遍历先保留的策略,所以SD算法处理后产生的间隙原子和OVITO处理后产生的间隙原子可能会出现位置相同但类型不同的情况,所以在评估SD算法的正确性的时候本文只对间隙原子的数量和对应格点的位置进行对比分析。由于体系中的空位是间隙原子进入具有原子的原胞造成的,所以间隙原子的数目等于空位的数目。
表 1和表 2表示在含2×106个原子的MD输出文件中,设置间隙原子的个数为4、20、50、100、200、400时,分别使用本文实现的MD与KMC耦合模拟的中间处理程序中和OVITO中的Wigner-Seitz原胞缺陷分析法处理后得到的间隙原子数和空位数。
表 1 SD算法处理结果和用OVITO中的Wigner-Seitz原胞缺陷分析法处理结果对比(间隙原子数量)
算法 间隙原子数量 SD 4 20 50 100 200 400 OVITO 4 20 50 100 200 400 表 2 SD算法处理结果和用OVITO中的Wigner-Seitz原胞缺陷分析法处理结果对比(空位数量)
算法 空位数量 SD 4 20 50 100 200 400 OVITO 4 20 50 100 200 400 -
本文实现的用MD和KMC耦合的方法模拟在温度为600 K时,Fe-0.3Cu(摩尔分数,%)的RPV (reactor pressure vessel)钢的辐照损伤实验。实验用MD方法模拟RPV钢中原子级联碰撞产生空位的过程;用KMC方法模拟团簇的形核与长大过程。模拟盒大小为100a×100a×100a(a为晶格常数,大小为0.285 53 nm),原子总数为2×106。模拟过程中采用周期性边界条件,计算所用势函数为EAM势。
本文用并行分子动力学模拟软件LAMMPS[13]使用MD方法模拟RPV钢的辐照损伤产生缺陷(主要是空位和间隙原子)的过程[14],模拟过程时间均是程序模拟材料演化的时间,而非计算机的运行时间,模拟时间为0.37 ps,在MD模拟结束时进行能量最小化处理。图 8为MD模拟结束后的原子可视图,其中,灰色圆点表示铜原子,白色圆点表示空位,黑色部分为铁原子。图 8b是图 8a中空位聚集部分对应的放大图。

图 8 MD模拟级联碰撞结果可视图
从图 8可以看出,在Fe-Cu合金中,通过级联碰撞产生了空位缺陷,缺陷的位置虽然比较集中(高能快中子打入造成的),但还都是离散的,并没有形成团簇。通过耦合程序的处理,对MD输出的原子进行清洗,除去间隙原子,并将原子映射到对应的格点位置,最后对数据进行格式转换处理,将体系格点处的原子或缺陷的排布传递给相应KMC模拟程序。程序接收原子和缺陷信息,并进入对缺陷在辐照条件下的形核与长大过程进行模拟。本文中KMC的模拟时间为1.25×10-2 mcs (mcs为KMC模拟程序输出的模拟时间单位,实际的时间单位秒可通过公式进行转换),模拟结束时的模拟结果如图 9所示。

图 9 KMC模拟形核与长大过程结果可视图
在图 9中,所有空位聚集在一起,形成一个大的空位团簇(空洞)。由于MD过程中由高能快中子打出的的空位分布过于集中,在KMC模拟过程中所有空位聚集到一起的所需时间较短。并且,单个空位跃迁经过的路径也较短,所以每个空位与周围金属原子交换位置的次数也就较少。这也就使得体系中虽有铜团簇形成(聚在一起的白色小球),但直径很小,且数量较少。上述结果符合辐照损伤的演化过程,说明MD与KMC耦合模拟以及整个并行耦合系统模拟是可行的。
-
本文采用MD和KMC耦合模拟的方法,对材料核辐照损伤过程中缺陷的产生以及形核和长大阶段进行模拟。通过对MD方法和KMC方法进行研究,确立了用MD和KMC方法耦合进行辐照损伤模拟的方案,即用MD方法模拟材料在持续的中子辐照下产生缺陷的过程,用KMC方法模拟缺陷形核和长大的过程。由于MD和KMC模拟中的原子表示及可模拟的原子类型不同,本文开发了一个耦合程序来实现MD和KMC的耦合模拟,在此过程中提出并实现了SD算法来完成原子与格点的映射,并在最后通过实验验证了SD算法以及MD和KMC耦合模拟的正确性和有效性。
Research and Implementation of Coupling Simulation between MD and KMC
-
摘要: 材料辐照损伤是当前材料领域和计算机领域研究的热点之一。分子动力学(MD)和动力学蒙特卡罗(KMC)耦合模拟是材料辐照损伤模拟中常用的方法。MD和KMC模拟体系中的原子类型、变量种类以及数据表示形式都不同,如何实现两个体系间数据的传输,是耦合模拟中的一个重要问题。为了解决这一问题,该文设计并开发了一个中间程序,提出了识别系统中原子类型的最短距离(SD)算法,通过计算原子位置与标准网格点之间的距离,来判断系统中的间隙原子、空位和正常原子。最后通过实验验证了该方法,MD及KMC耦合模拟的正确性和有效性。Abstract: The radiation damage has become a hot focus in materials and computer field. Molecular dynamics method (MD) and kinetic Monte Carlo method (KMC) are two commonly used methods to study the phenomenon. Due to the MD and KMC simulation system have different atom types, variables and data representations. It is an important issue to realize the data transmission between the two systems. Therefore, we develop a middle program to build a bridge between the two simulations, and puts forward shortest distance algorithm (SD) which can identify the type of atom in the system. The SD algorithm identifies the type of atom in the system by calculating the distance between the atomic location and the standard grid point. Finally, this paper verifies the correctness and effectiveness of the SD algorithm and coupling simulation between MD and KMC.
-
Key words:
- coupling simulation /
- KMC /
- MD /
- shortest distance algorithm
-
表 1 SD算法处理结果和用OVITO中的Wigner-Seitz原胞缺陷分析法处理结果对比(间隙原子数量)
算法 间隙原子数量 SD 4 20 50 100 200 400 OVITO 4 20 50 100 200 400  下载: 导出CSV
下载: 导出CSV
表 2 SD算法处理结果和用OVITO中的Wigner-Seitz原胞缺陷分析法处理结果对比(空位数量)
算法 空位数量 SD 4 20 50 100 200 400 OVITO 4 20 50 100 200 400
下载: 导出CSV
-
[1] 林江宏, 林锦贤, 吕暾.多核CPU和GPU加速分子动力学模拟[J].计算机应用, 2011, 31(3):843-847. http://www.cqvip.com/QK/94832X/201103/36682443.html LIN Jiang-hong, LIN Jin-xian, LÜ Tun. Accelerated molecular dynamics simulation using multi-core CPU and GPU[J]. Journal of Computer Applications, 2011, 31(3):843-847. http://www.cqvip.com/QK/94832X/201103/36682443.html [2] XU H, STOLLER R E, BÉLAND L K, et al. Self-evolving atomistic kinetic Monte Carlo simulations of defects in materials[J]. Computational Materials Science, 2015, 100:135-143. doi: 10.1016/j.commatsci.2014.12.026 [3] VOTER A F. Introduction to the kinetic Monte Carlo method[M]//SICKAFUS K E, KOTOMIN E A, UBERUAGA B P. Radiation Effects in Solids. Netherlands, Springer, 2007: 1-23. [4] WARRIER M, BHARDWAJ U, BUKKURU S. Multi-scale modeling of radiation damage: Large scale data analysis[J]. 2016, 759(1): 12-78. http://www.researchgate.net/publication/309963553_Multi-scale_Modeling_of_Radiation_Damage_Large_Scale_Data_Analysis [5] 唐杜, 贺朝会, 臧航, 等.硅单粒子位移损伤多尺度模拟研究[J].物理学报, 2016, 65(8):193-200. http://www.cnki.com.cn/Article/CJFDTotal-QJGY201511003.htm TANG du, HE Chao-hui, ZANG Hang, et al. Multi-scale simulations of single particle displacement damage in silicon[J]. Acta Physica Sinica, 2016, 65(8):193-200. http://www.cnki.com.cn/Article/CJFDTotal-QJGY201511003.htm [6] ARISTOFF D, CHILL S, SIMPSON G. Analysis of estimators for adaptive Kinetic Monte Carlo[J]. Mathematics, 2015, 18(4):546-553. http://www.researchgate.net/publication/278734121_Analysis_of_estimators_for_adaptive_Kinetic_Monte_Carlo [7] NEUMANN P, BIAN X. MAMICO:Transient multi-instance molecular-continuum flow simulation on supercomputers[J]. Computer Physics Communications, 2017, 220:390-402. doi: 10.1016/j.cpc.2017.06.026 [8] MOLNAR D, MUKHERJEE R, CHOUDHURY A, et al. Multiscale simulations on the coarsening of Cu-rich precipitates in α-Fe using kinetic Monte Carlo, molecular dynamics and phase-field simulations[J]. Acta Materialia, 2012, 60(20):6961-6971. doi: 10.1016/j.actamat.2012.08.051 [9] CATURLA M J, RUBIA T D D L, VICTORIA M, et al. Multiscale modeling of radiation damage:Applications to damage production by GeV proton irradiation of Cu and W, and pulsed irradiation effects in Cu and Fe[J]. Journal of Nuclear Materials, 2001, 296(1):90-100. http://www.sciencedirect.com/science/article/pii/S0022311501005694 [10] MORISHITA K, SUGANO R, WIRTH B D. MD and KMC modeling of the growth and shrinkage mechanisms of helium-vacancy clusters in Fe[J]. Journal of Nuclear Materials, 2003, 323(2-3):243-250. doi: 10.1016/j.jnucmat.2003.08.019 [11] STUKOWSKI A. Visualization and analysis strategies for atomistic simulations[M]//WEINBERGER C, TUCKER G. Multiscale Materials Modeling for Nanomechanics. [S.l.]: Springer, 2016: 317-336. [12] ASHCROFT N W, MERMIN N D. Solid state physics[M]. Philadelphia:Saunders Company, 1976. [13] JEFFERS J, REINDERS J, SODANI A. Intel Xeon Phi processor high performance programming[M]. San Francisco:Morgan Kaufmann Publishers Inc, 2016:443-470. [14] THOMPSON A P. Predictive atomistic simulations of materials using LAMMPS[R]. Albuquerque, NM(United States): Sandia National Lab, 2016. https://www.osti.gov/biblio/1394896 -
 点击查看大图
点击查看大图

图(9) / 表(2)
计量
- 文章访问数: 4152
- HTML全文浏览量: 1804
- PDF下载量: 183
- 被引次数: 0
