 ISSN
ISSN 



-
人类第三代测序技术的迅速发展,让生命系统组成元件间的相互作用关系信息得到更加快速的积累。基因数据的不断丰富,表型数据的不断增加,为理解疾病与致病基因之间的关系提供了大量有效的数据。在生物数据大量涌现的前提下,利用相关计算技术和模型对数据进行分析与挖掘,加快了生物学研究前进的步伐,可以深层次挖掘疾病表型与致病基因之间的关系,为了解疾病发病机理、疾病临床诊断和疾病预防与治疗提供了便利。
通过几十年的努力,人类已经发现了一些疾病的致病基因,如BRCA1和BRCA2基因在乳腺癌的发生中发挥重要的作用[1],EGFR在肺癌的发生中发挥重要作用[2]。如果能够知道更多疾病的致病基因,则可以在发病前期进行基因检测预防,在发病过程中进行相应的治疗,后续也可以将发病机理应用到药物设计中,从而有效提高疾病的控制与治愈能力。通过疾病表型和致病基因关系的挖掘,使得疾病发病机理一目了然,在疾病发现过程中能直击疾病发病原因,后续治疗能做到药到病除。
-
目前,挖掘疾病表型与致病基因的关联关系是一个极具挑战的课题。如果能够设计出高精度的致病基因预测方法,对于生物学家、临床医师和遗传学家等相关人员来说具有非常重要的意义。这不但有助于提高发现致病基因的准确率,缩短发现致病基因的周期,节省大量的人力物力,同时也为将来的生物医学和基因治疗诊断等技术的发展奠定重要基础。
随着计算机和生物技术的迅猛发展,大量的生物信息数据的产生,疾病和基因知识的可用性大幅度提高,科研人员也相应提出了一系列疾病与基因预测的计算方法。其中,随机游走是疾病与基因关联关系预测中较为常见的办法,主要包括重启随机游走和双向随机游走等几种类型。文献[3]在双层耦合网络上提出了重启随机游走,用于推断潜在的miRNA与疾病的相关性。文献[4]开发了BiRWHMDA的计算模型,通过在双层耦合网络上的双向随机游走来预测潜在的微生物与疾病关联。文献[5]提出在双层耦合网络上基于多路径的双向随机游走预测微生物与疾病相关性。文献[6]结合表型相似网络、基因相似网络和表型基因关联网络构成表型基因双层耦合网络,并在其上采用重启随机游走算法,推出了一种新的预测疾病致病基因的方法。文献[7]采用了带重启的随机游走算法和最短路径这两种广泛使用的算法,构造了两种参数化计算方法,即基于RWR的方法和基于SP的方法,并在此基础上构建了一种新的疾病基因识别的集成方法。
利用矩阵预测疾病与基因关系也是一个不错的办法。文献[8]提出了一种基于归纳式矩阵补全预测潜在lncRNA与疾病相关性的方法(predict lncRNA-disease associations from known data using IMC, SIMCLDA)。文献[9]开发了一种利用协同矩阵因子分解预测人类微生物疾病相关性的模型(collaborative matrix factorization for human microbe-disease association, CMFHMDA)。文献[10]提出一种基于Katz方法的预估计和基于归纳型矩阵补全方法的精化估计两步骤的Katz增强归纳型矩阵补全的基因−疾病关联预测模型。
把高斯相互作用应用于预测之中,文献[11]应用高斯相互作用轮廓核相似测度确定微生物相似性和疾病相似性。文献[12]建立了用于miRNAs与疾病相关性预测的双层耦合网络推理的计算模型,通过整合miRNAs功能相似性、疾病语义相似性、高斯相互作用来揭示潜在的miRNAs与疾病相关性。
将路径作为预测分数,文献[13]引入PBHMDA(path-based human microbe-disease association),通过对微生物与疾病之间的所有路径进行评估,得出每个候选微生物与疾病对的预测得分。
研究人员还提出了其他一些疾病与基因关系预测的办法。文献[14]提出了一种基于SimRank和密度聚类推荐模型的miRNA与疾病相关性预测方法(based on the SimRank and density-based clustering recommender model for miRNA-disease associations prediction, SRMDAP)。文献[15]基于miRNA与疾病关联预测评分模型(within and between score for MiRNA-disease association prediction, WBSMDA)预测与各种复杂疾病关联的miRNAs。文献[16]采用拉普拉斯正则化最小二乘分类器(Laplacian regularized least squares for human microbe–disease association, LRLSHMDA)建立预测模型。文献[17]将链路预测的思想引入到长非编码RNA−疾病关联预测中。文献[18]提出一种基于密度聚类的二分网络投影算法(bipartite network projection based on density clustering to predict miRNA-disease associations, BNPDCMDA)来预测miRNA−疾病关联。
以随机游走为主导思想的预测方法能够扩大候选基因的范围,可以避免遗漏连接度低和网络边缘的节点,尤其是在多基因疾病的预测中,可以大大提高预测候选致病基因方法的性能;在矩阵预测中,数据的稀疏对预测有很大的影响,PU问题也是需要面对的另一个问题,加入Katz方法也只缓解部分影响;使用高斯相互作用预测将疾病或者基因的相互作用信息作为特征向量,引入高斯核函数,计算疾病或基因间的相似度后在进行疾病和基因之间的相似预测,但是对高斯相互作用相似度参数标准化后,基因或疾病高斯核相互作用相似值就不在依赖于数据集;路径预测利用了生物信息节点之间的拓扑结构,在拓扑结构的基础上预测;其他一些算法都是基于机器学习的一些思想进行关联预测的,然而有监督的机器学习算法,需要假设与疾病相关的基因和不相关的基因是不关联的,但是被证明与疾病相关的基因数量较少,且很少有实验能够证明那些关系是不存在的。
进行多种算法比较研究后,可知基于随机游走的方法相比矩阵预测或聚类的方法存在一定优越性。本文根据疾病表型和疾病基因数据节点属于不同类型节点这一特点,基于疾病表型和疾病基因数据来构成双层耦合网络,提出了在表型−基因的双层耦合网络基础上进行带有元路径的随机游走,从而实现关联关系的预测与分析算法。
-
复杂网络的研究大多局限于单个网络,而事实上单个网络仅仅是更大复杂系统中的一个子集,复杂系统往往是由许多具有不同结构与功能的网络耦合而成的[19]。多层耦合网络由多个子网络构成,网络中每一层通过一些共享节点而耦合在一起,各层的节点具有不同的属性,并且各层之间的节点存在耦合关系,一般分为相互依赖和相互协作两种关系。例如,在线购物交易平台依赖于因特网,因特网又依赖于电力网;公路网和铁路网组成的双层协作网络,两者相互协作保障了人们出行的方便快捷。作为结果,一个网络中的信息传播可能出现在另一个网络扩散,并最终导致一个信息级联效应。
本文利用小鼠的已知疾病表型之间的关联关系、已知致病基因之间的关联关系和已知疾病表型与致病基因之间的关联关系,构建出表型−基因的双层耦合网络。在表型−基因的双层耦合网络中,上层为表型关联网络,下层为基因关联网络,上下网络之间通过表型与基因的关联关系进行耦合。
-
信息网络[20]是一个带有对象类型的映射函数
$\tau :{\cal{V}} \to {\cal{A}}$ 和链接类型映射函数$\phi :{\cal{E}} \to {\cal{R}}$ 的图$G = $ $({\cal{V}},{\cal{E}}) $ ,其中每个对象$v \in {\cal{V}}$ 属于一个特定的对象类型$\tau (v) \in {\cal{A}}$ ,每个链接$e \in \varepsilon $ 属于一个特定的关系$\phi (e) \in {\cal{R}}$ ,如果两个链接属于同一个关系类型,那么这两个链接具有相同类型的开始对象和结束对象。 -
表型关联网络是一种信息网络,可以定义为
${N_P} = \left( {P,{E_{PP}},{W_{PP}}} \right)$ ,其中$P = \left\{ {{p_1},{p_2}, \cdots ,{p_m}} \right\}$ 表示表型节点的集合,${E_{PP}}$ 表示表型之间的关联关系,${W_{PP}}$ 表示关联关系权重值,如果表型$i$ 与表型$j$ 有关联关系,则权重值为1,否则为0。表示如下:本文中表型关联网络需要的数据从MGI数据库中获取得到,表型关联网络示意图如图1所示。

-
基因关联网络定义为
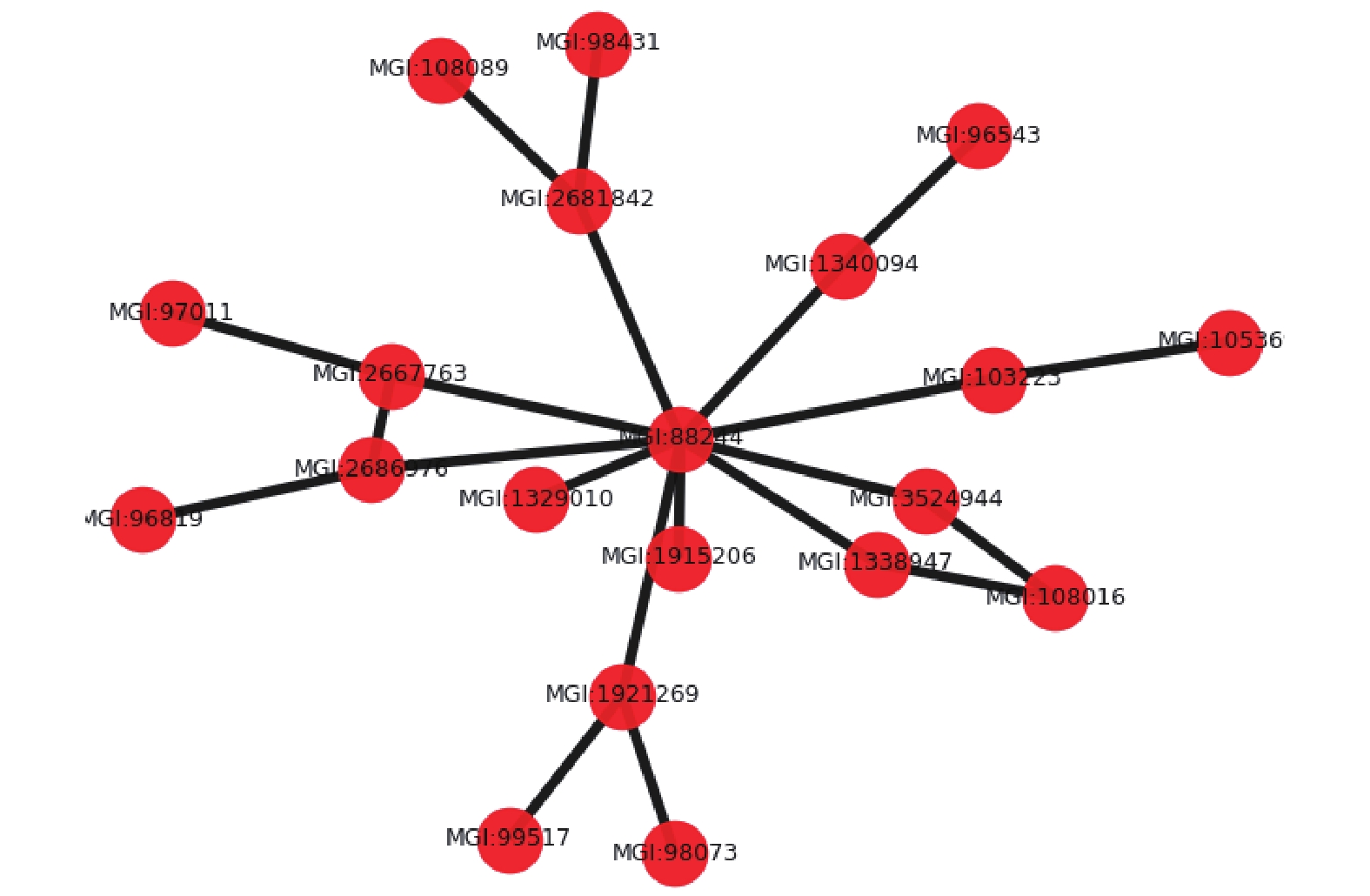
${N_G} = \left( {G,{E_{GG}},{W_{GG}}} \right)$ ,其中$G = \left\{ {{{{g}}_1},{{{g}}_2}, \cdots ,{{{g}}_n}} \right\}$ 表示基因节点的集合,${E_{GG}}$ 表示基因之间的关联关系,${W_{GG}}$ 表示关联关系权重值,基因$i$ 与基因$j$ 有关联关系则权重值为数据库中所给数值,用$\alpha $ 表示,否则为0。表示如下:文中基因关联网络需要的数据从MouseNet下载,基因关联网络示意图如图2所示。

-
表型−基因网络数据来源于MGI数据库,定义为:
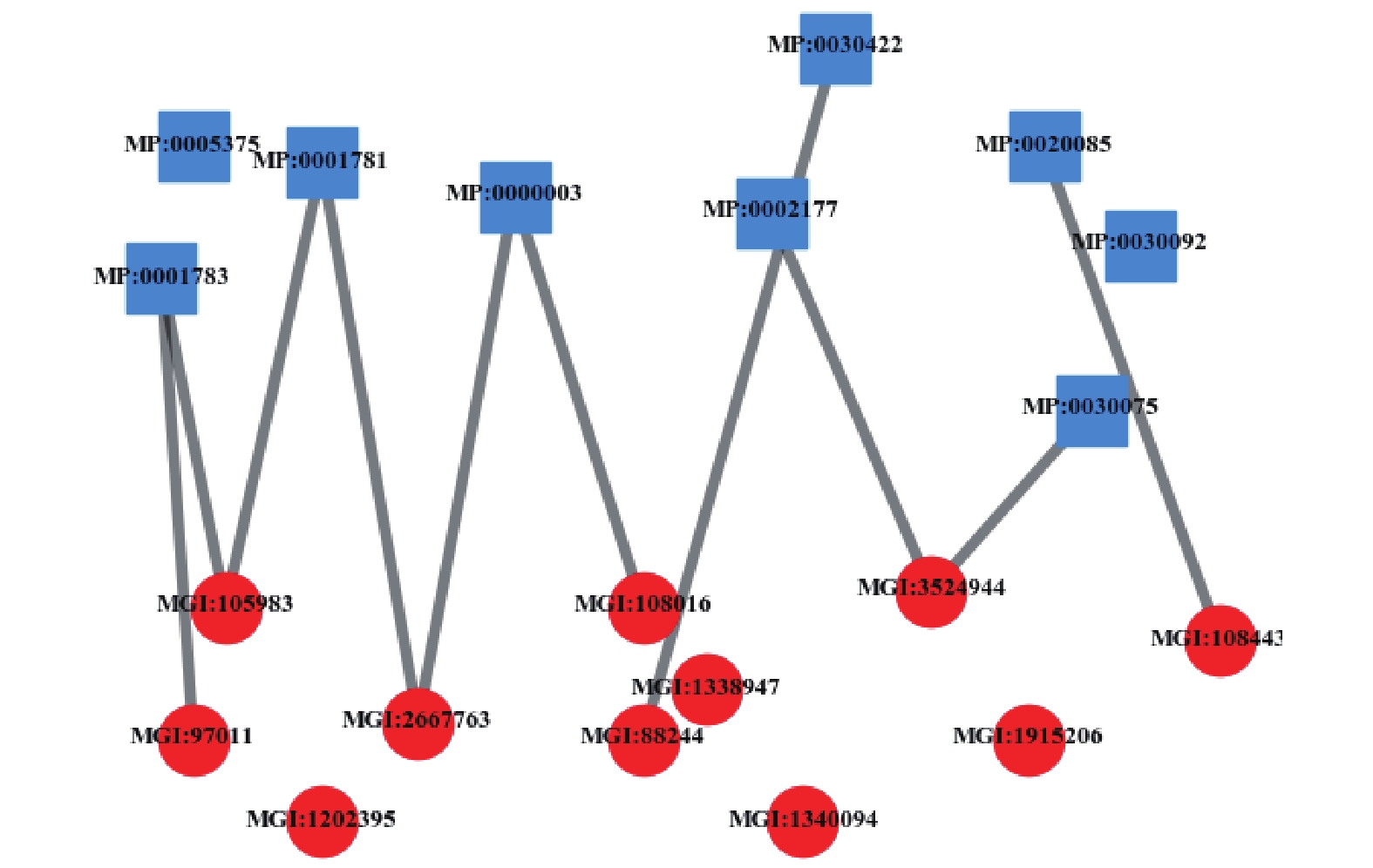
${N_{PG}} = \left( {P \cup G,{E_{PG}},{W_{PG}}} \right)$ ,其中:$P \cup G = \{ {p_1},{p_2}, \cdots ,$ ${p_m},{g_1},{g_2}, \cdots ,{g_n}\}$ 表示表型和基因节点的集合,${E_{PG}}$ 表示表型与基因之间的关联关系,${W_{PG}}$ 表示关联关系权重值,如果表型$i$ 与基因$j$ 有关联关系则权重值为1,否则为0。表示如下:表型−基因关联网络示意图如图3所示。

-
表型−基因双层耦合网络就是在表型关联网络
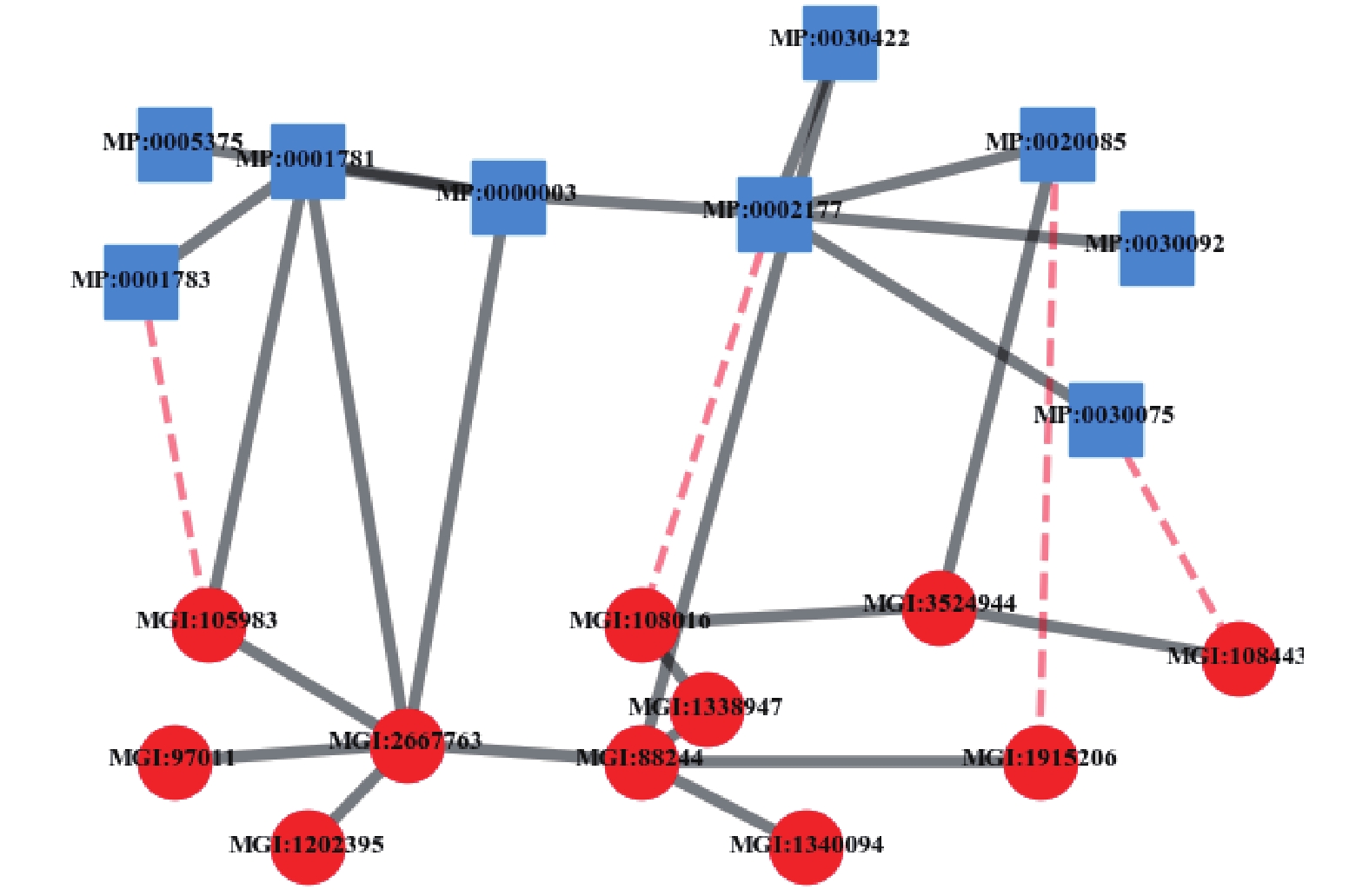
${N_P}$ 、基因关联网络${N_G}$ 和表型−基因关联网络${N_{PG}}$ 基础上,上层为表型网络${N_P}$ ,下层为基因网络${N_G}$ ,表型−基因关联网络${N_{PG}}$ 节点间的关系作为上下层间的耦合关系而得到,可以定义为:${N_{P - G}} = ( V = P \cup G, $ $E = {E_{PP}} \cup {E_{PG}} \cup {E_{GG}},W = {W_{PP}} \cup{W_{PG}} \cup {W_{GG}})$ ,其中$V = P \cup G$ 表示包括表型与基因的所有节点,$E = {E_{PP}} \cup {E_{PG}} \cup {E_{GG}}$ 表示节点间的链接关系,$W = {W_{PP}} \cup {W_{PG}} \cup {W_{GG}}$ 表示节点链接关系的权重值,表型−基因双层耦合网示意图如图4所示。
图4中,实线部分为已知存在的关联关系,包括了表型与表型的关联、基因与基因的关联和表型与基因的关联;虚线部分为待预测的表型与基因的关系是否关联。
-
在2.1节定义的基础上,如果对象类型
$|{\cal{A}}| > 1$ 或者关系类型$|{\cal{R}}| > 1$ 时,该信息网络为异构信息网络。从图4中可以看出在表型−基因双层耦合网${N_{P - G}} = \left( {V = P \cup G,E = {E_{PP}} \cup {E_{PG}} \cup {E_{GG}},W = } \right.{W_{PP}} \cup $ $ {W_{PG}}\cup {W_{GG}})$ 中,表型关联网络${N_P}$ 和基因关联网络${N_G}$ 的节点分属两个类型,通过表型−基因关联网络${N_{PG}}$ 进行耦合,整体上看表型−基因双层耦合网为一个异构网络。 -
元路径(meta-path)[20]主要用来描述异构网络中任意两个节点间的不同路径类型,可以定义为:在带有对象类型映射
$\tau :{\cal{V}} \to {\cal{A}}$ 和链接类型映射$\phi :{\cal{E}} \to {\cal{R}}$ 的异构网络$G = ({\cal{V}},{\cal{E}})$ 的元模板上的一条路径,其形式为${A_1}\mathop \to \limits^{{R_1}} {A_2}\mathop \to \limits^{{R_2}} \cdots \to {A_{l + 1}}$ 。元路径${A_1}\mathop \to \limits^{{R_1}} {A_2}\mathop \to \limits^{{R_2}} \cdots \to {A_{l + 1}}$ 描述了类型${A_1}$ 到类型${A_{l + 1}}$ 间的复合关系$R = {R_1} \circ {R_2} \circ \cdots \circ {R_l}$ ,其中“$ \circ $ ”表示关系上的复合运算。在表型−基因双层耦合网络
${N_{P - G}}$ 中两个节点之间就存在不同类型不同长度的元路径,以图4为例,可以有$P \to P \to G$ 、$P \to P \to G \to G$ 、$P \to P \to $ $P \to P \to G $ 等。对于一个给定的网络,可能存在的元路径数目与路径长度成指数增长[21]。选择不同的元路径,表型与基因之间的关联性也不同,同时,文献[20]指出很长的元路径并不是很有意义,反而路径长度越大,关系越弱,预测也越模糊。因此,在表型与基因的关联预测中,本文主要考虑如下4条元路径,如表1所示。序号 元路径 $M{P_1}$ $P \to P \to G$ $M{P_2}$ $P \to G \to G$ $M{P_3}$ $P \to P \to G \to G$ $M{P_4}$ $P \to G \to P \to G$ -
随机游走(random walk)又称随机游动或随机漫步,是一种数学统计模型,在金融、物理和社交媒体等复杂网络分析中都有广泛应用。随机游走模型是从图上一个或一组节点开始,通过迭代随机的访问图中的每一个节点。每一次移动时,当前节点都以一定的概率移动到他们的邻居节点。因此,图中每个节点都会获得一个经计算得到的当前节点游走到该节点的概率分布值[22]。文献[23]提出了基于双层耦合网络的随机游走RWRH算法。RWRH算法在不同的网络中游走,从网络
${G_1}$ 或者网络${G_2}$ 的某一节点开始进行随机游走,在游走过程中,以一定的概率停留在网络${G_1}$ 的下一个节点或者网络${G_2}$ 的一个节点。在表型−基因双层耦合网络
${N_{P - G}}$ 中选定了元路径,随机游走将基于元路径进行游走,但是,游走到元路径中指定类型节点中的哪一个节点是未知的,即规定了下一步游走的节点类型但不固定某个节点。那么,表型−基因双层耦合网络${N_{P - G}}$ 中节点在既定的元路径$P \to P \to G$ 、$P \to G \to G$ 、$P \to P \to G \to G$ 和$P \to G \to P \to G$ 下由上一个节点游走到下一个节点的跳转概率有如下4种表示:式中,
$i$ 表示第$i$ 步跳转。将上式用矩阵形式表示如下:
1)当
${v^i} \in P\text{,}{v^{i + 1}} \in P$ ,则一步跳转概率矩阵为${ D}_{PP}^{ - 1}{{ W}_{PP}}$ ;2)当
${v^i} \in P\text{,}{v^{i + 1}} \in G$ ,则一步跳转概率矩阵为${ D}_{PG}^{ - 1}{{ W}_{PG}}$ ;3)当
${v^i} \in G\text{,}{v^{i + 1}} \in P$ ,则一步跳转概率矩阵为${ D}_{GP}^{ - 1}{{ W}_{GP}}$ ,${{ W}_{GP}} = { W}_{PG}^{\rm T}$ ;4)当
${v^i} \in G\text{,}{v^{i + 1}} \in G$ ,则一步跳转概率矩阵为${ D}_{GG}^{ - 1}{{ W}_{GG}}$ 。其中,
${{ D}_{PP}}$ 、${{ D}_{PG}}$ 、${{ D}_{GP}}$ 、${{ D}_{GG}}$ 为对角矩阵,对角线上的值分别为${{ W}_{PP}}$ 、${{ W}_{PG}}$ 、${{ W}_{GP}}$ 、${{ W}_{GG}}$ 中对应行元素之和,即:因此,在表型-基因双层耦合网络
${N_{P - G}} = ( V = P \cup G,$ $E = {E_{PP}} \cup {E_{PG}} \cup {E_{GG}},W = {W_{PP}} \cup {W_{PG}} \cup {W_{GG}})$ 中,基于元路径$M{P_1}:P \to P \to G$ 的表型${p_i}$ 到基因${g_i}$ 的跳转概率矩阵${{ X}_{PPG}}$ 可表示为:基于元路径
$M{P_2}:P \to G \to G$ 的表型${p_i}$ 到基因${g_i}$ 的跳转概率矩阵${{ {X}}_{PGG}}$ 可表示为:基于元路径
$M{P_3}:P \to P \to G \to G$ 的表型${p_i}$ 到基因${g_i}$ 的跳转概率矩阵${{ X}_{PPGG}}$ ,可以表示为:基于元路径
$M{P_4}:P \to G \to P \to G$ 的表型${p_i}$ 到基因${g_i}$ 的跳转概率矩阵${{ X}_{PPGG}}$ 可表示为: -
对于表型−基因双层耦合网络
${N_{P - G}} = ( V = P \cup G, $ $E = {E_{PP}} \cup {E_{PG}} \cup {E_{GG}},W = {W_{PP}} \cup{W_{PG}} \cup {W_{GG}})$ 中的任意的表型${p_i}$ 和基因${g_j}(i = 1,2, \cdots ,m;j = 1,2, \cdots ,n)$ ,如果$( {{p_i},{{\bf{g}}_j}} ) \notin {E_{PG}}$ 或者${W_{PG}}(i,j) = 0$ ,则需要对其关联性进行预测。在综合不同元路径的情况下,按不同元路径所占权重进行累加,可以得到表型
${p_i}$ 到基因${g_j}$ 之间的跳转概率矩阵X:在得到的跳转概率矩阵X中,其对应的取值就是表型
${p_i}$ 到基因${g_i}$ 的关联值大小,值越大,关联越紧密;反之亦然。 -
MGI是实验室小鼠的国际数据库资源,包含:小鼠基因组数据库(MGD)、基因表达数据库(GXD)、小鼠肿瘤生物学(MTB)数据库、基因本体(GO)项目等。本文用到的表型数据和表型-基因数据集从MGI数据库资源下载获得。其中,表型数据集包含了12 838个疾病表型,构成了16 108对表型与表型关联对;表型-基因数据集共有表型与基因的关联数据对37 246对。
MouseNet V2是许多生物医学研究选择的一种改进的实验小鼠功能基因网络。MouseNet V2为2008年MouseNet的改进版本,加入了大量来自不同生物的新微阵列数据。MouseNet V2现在覆盖88%的编码基因组,具有更高的准确性。本文使用基因数据即从MouseNet V2中获得,共有17 710个基因,构成了关联基因对788 081对。
-
在4条元路径
$M{P_1}:P \to P \to G$ 、$M{P_2}:P \to $ $G \to G$ 、$M{P_3}:P \to P \to G \to G$ 和$M{P_4}:P \to G \to $ $P \to G$ 中进行随机游走得到了表型在4条元路径下游走到基因的跳转概率矩阵,即${{ X}_{PPG}}$ 、${{ X}_{PGG}}$ 、${{ X}_{PPGG}}$ 和${{ X}_{PGPG}}$ 。在所得到的${{ X}_{PPG}}$ 、${{ X}_{PGG}}$ 、${{ X}_{PPGG}}$ 和${{ X}_{PGPG}}$ 数据中,找出4个数据都同时存在的表型到基因的概率,在此前提下使用主成分分析的办法,即通过变量变换的方法把相关的变量变为若干不相关的综合指标变量,从而实现对数据集的降维,在过程中求出综合评价函数而得到不同元路径下的权重值,即是${ X} = {\alpha _{PPG}}{{ X}_{PPG}} + {\alpha _{PGG}}{{ X}_{PGG}} +{\alpha _{PPGG}}{{ X}_{PPGG}} + $ $ {\alpha _{PGPG}}{{ X}_{PGPG}}$ 中${\alpha _{PPG}}$ 、${\alpha _{PGG}}$ 、${\alpha _{PPGG}}$ 和${\alpha _{PGPG}}$ 的值。最后进行表型到基因在元路径下按权重累加,并选出前$k$ 名为最终结果,作为表型与基因关联关系的预测值。 -
为了评价本文算法预测表型与基因关联关系的性能,采用留一交叉验证法(leave-one-out cross validation, LOO)实验。在数据的N个样本中,每次实验将一个样本作为测试集,剩下的N−1个样本作为训练集,直到所有的样本都被作为测试集,即得到N个模型,在此过程中利用接收者操作特征(ROC)曲线[24]对预测性能进行评价,绘制截止时的真阳性率(TPR、敏感性或召回)与假阳性率(FPR、1-特异性)的关系曲线。
在ROC曲线绘制和AUC面积的计算时,使用到如下的定义:
其中,条件正(P):数据中实际正案例数;条件负(N):数据中的实际负案例数;TP和TN代表正确预测的真正和真负数量;FP和FN代表错误预测的假阳性和假阴性。
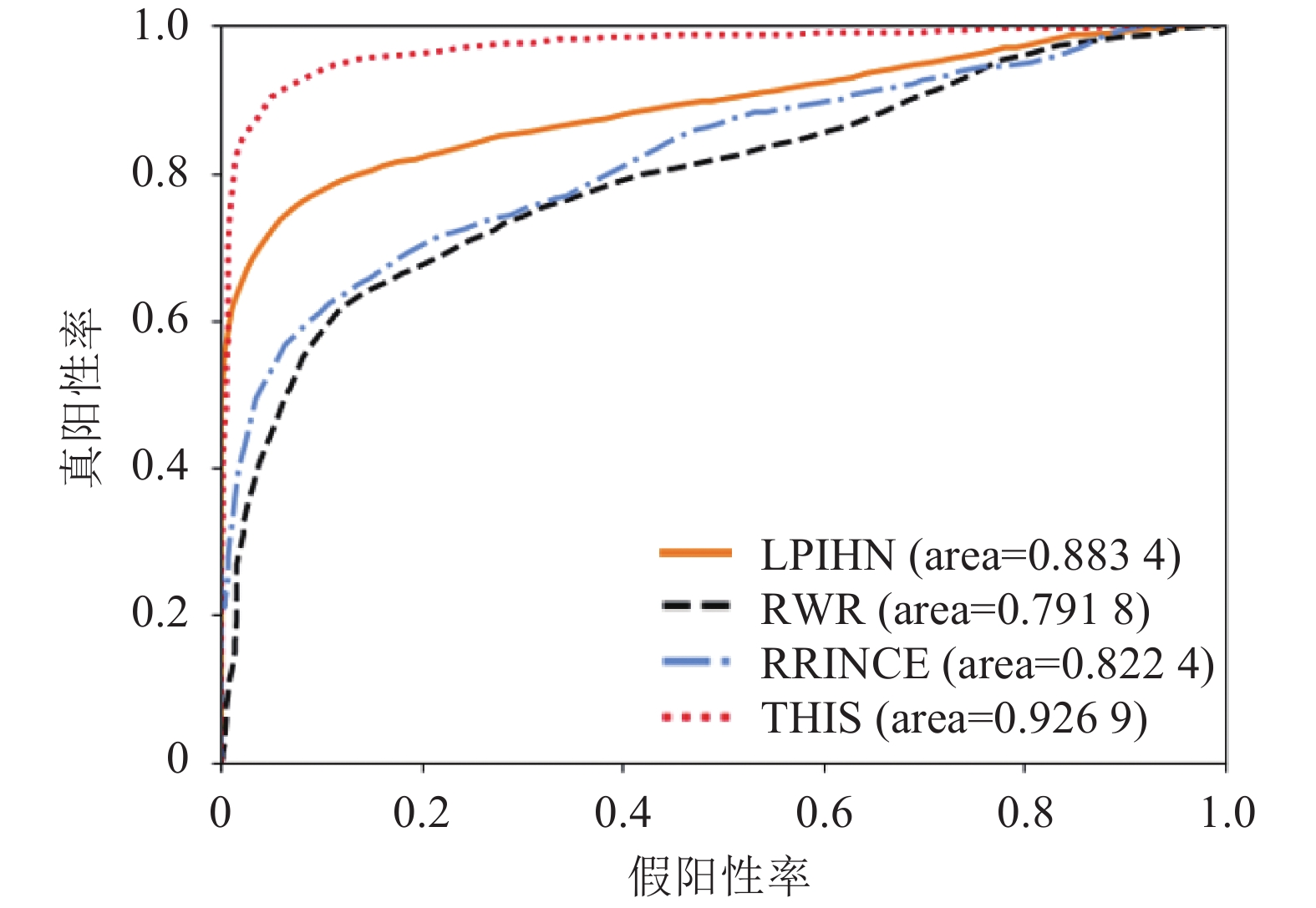
将本文算法与其他3种相关预测算法RWR[25]、LPIHN[26]和PRINCE[27]进行测试比较。RWR算法从已知的致病基因以相同的概率出发,随机走向邻居节点,当前后两次游走的概率向量相同或者前后两次游走的概率差值小于某个阀值时,认为游走达到平衡,然后将概率值从大到小排序,排名靠前的说明基因与疾病的相关性较大,认为该基因是该疾病的致病基因。LPIHN是一种在异构网络上实现随机游走的方法。PRINCE是一种基于对优先级函数的约束的全局方法,从某个查询疾病表型出发游走至整个网络,通过计算在基因节点邻居中与查询疾病关联的基因的优先次序后,合并相似性信息中分数高的基因作为致病基因。RWR方法中的重启概率
$r$ 经过多次试验,对试验结果影响不大,所以设置$r = 0.5$ ;LPIHN的参数根据[26]文中提及参数值特设置如下:$\gamma = 0.5$ ,$\beta = 0.5$ ,$\delta = 0.3$ ;PRINCE的参数根据[27]文中提及数值而设置如下:$\alpha = 0.5$ ,$c = - 15$ ,$d = \lg (9\;999)$ ,传播迭代次数为10。所得结果如图5所示,其中THIS代表本文提出的算法。
结果表明,在所给数据实验中,本文提出的算法的AUC得分为93%,高于RWR、LPIHN和PRINCE的AUC值,分别为79%、88%和82%。
-
随着基因数据和表型数据的不断增加,为理解疾病与致病基因之间的关系提供了大量有效的数据,也为利用数据分析与挖掘的手段找出疾病表型与致病基因之间的关系提供了便利。为此,旨在设计一种算法来找到表型节点与基因节点的更多关联关系。本文在经典的随机游走方法上加入了元路径的概念,充分利用先验知识及网络中包含的生物关系来预测发现表型与基因的关联关系。从实验结果可以看出,本文算法的正确率高于RWR、LPIHN和PRINCE等算法,能够得到较好的预测效果。
在后续的工作中,有如下几方面可以做进一步研究:1) 整合更可靠的生物网络数据。生物信息知识的缺乏和实验数据的假阳性都会对实验的预测结果造成误差,整合其他有用的生物数据将会提高生物网络数据的可靠性。2) 整合多重生物网络数据。如将序列相似性、功能注释、微阵列表达、蛋白质域、通路成员等数据库整合为一个完整数据进行相应的预测。3) 改变生物网络的拓扑特征。可以适当改变网络的拓扑特征,如介数中心性、紧密中心性、聚类系数等,再进行关联预测。
Phenotype-Gene Association Analysis and Prediction Based on Double-Layer Coupled Network
doi: 10.12178/1001-0548.2019133
- Received Date: 2019-06-03
- Rev Recd Date: 2019-11-07
- Available Online: 2020-05-28
- Publish Date: 2020-05-01
-
Key words:
- correlation /
- disease phenotype /
- double-layer coupled network /
- pathogenic gene
Abstract: With the completion of genome sequencing and the continuous development of gene technology, the pathogenic genes of some diseases are gradually identified. At present, people have grasped the pathogenic causes of some diseases through scientific experiments, but the pathogenic causes of most diseases, especially those related to genes, are still unknown. In this paper, the mouse data with 85% homology similarity to human is used as the research object. The disease phenotype data set, pathogenic gene data set and confirmed phenotype-gene association data set are constructed into a double-layer coupled network. The data are analyzed and mined by meta-path random walk method, and the uncertainties are predicted on the basis of confirmed phenotype-gene association data. The proposed algorithm achieves better prediction results compared with other algorithms.
| Citation: | YU Yong, GU Jie, ZHAO Na, LUO Yong-jun, KAN Shi-lin. Phenotype-Gene Association Analysis and Prediction Based on Double-Layer Coupled Network[J]. Journal of University of Electronic Science and Technology of China, 2020, 49(3): 438-444. doi: 10.12178/1001-0548.2019133

|



















































































































































 DownLoad:
DownLoad: